For video accompanying this chapter see ExpertConsult.com . See inside cover for access details.
For video accompanying this chapter see ExpertConsult.com . See inside cover for access details.
Embryology
The primary and secondary palates arise from the frontonasal process and palatal processes of the maxilla, respectively, separated by the incisive foramen. From the incisive foramen, fusion of these processes occurs, progressing anteriorly during weeks 5–6 for the primary palate and posteriorly in weeks 7–8 for the secondary palate. Aberration of this process results in clefts between these embryologically distinct regions. More specifically, a cleft of the primary palate occurs between the premaxilla and ipsilateral palatal process of the maxilla, while a cleft of the secondary palate occurs between the two palatal shelves, with each shelf united to the premaxilla.
Cleft palate can occur in isolation or in combination with cleft lip, unilaterally or bilaterally. A cleft of the lip involving the primary palate terminates at the incisive foramen, in keeping with the embryological origin of these structures as previously described. When a cleft of the lip and primary palate occurs in combination with a cleft of the secondary palate, the latter two malformations may be in continuity, resulting in the classic unilateral or bilateral cleft lip and palate or discontinuous (for example, cleft lip and primary palate with cleft of the soft palate or incomplete cleft lip and complete cleft of the secondary palate) ( Fig. 17.1 ). An isolated cleft palate is subclassified as complete or incomplete, with the latter involving the soft palate with or without a portion of the posterior hard palate. In isolated cleft palate, the vomer is usually midline, vertically oriented, and attached to the palatal shelves as far back as the anterior extent of the cleft. The vomer also maintains a vertical orientation in complete bilateral cleft lip and palate; however, it may be laterally deviated contingent on the orientation of the premaxilla from which the vomer projects. This is in contradistinction to the morphology of the vomer in unilateral cleft lip and palate, where the cranial portion of the vomer is almost vertical with angulation (often at 90 degrees) of the caudal segment towards the palatal shelf to which it is attached on the non-cleft side. The point of attachment between vomer and palatal shelf can be discerned by the color variance of nasal and oral mucosa, respectively.
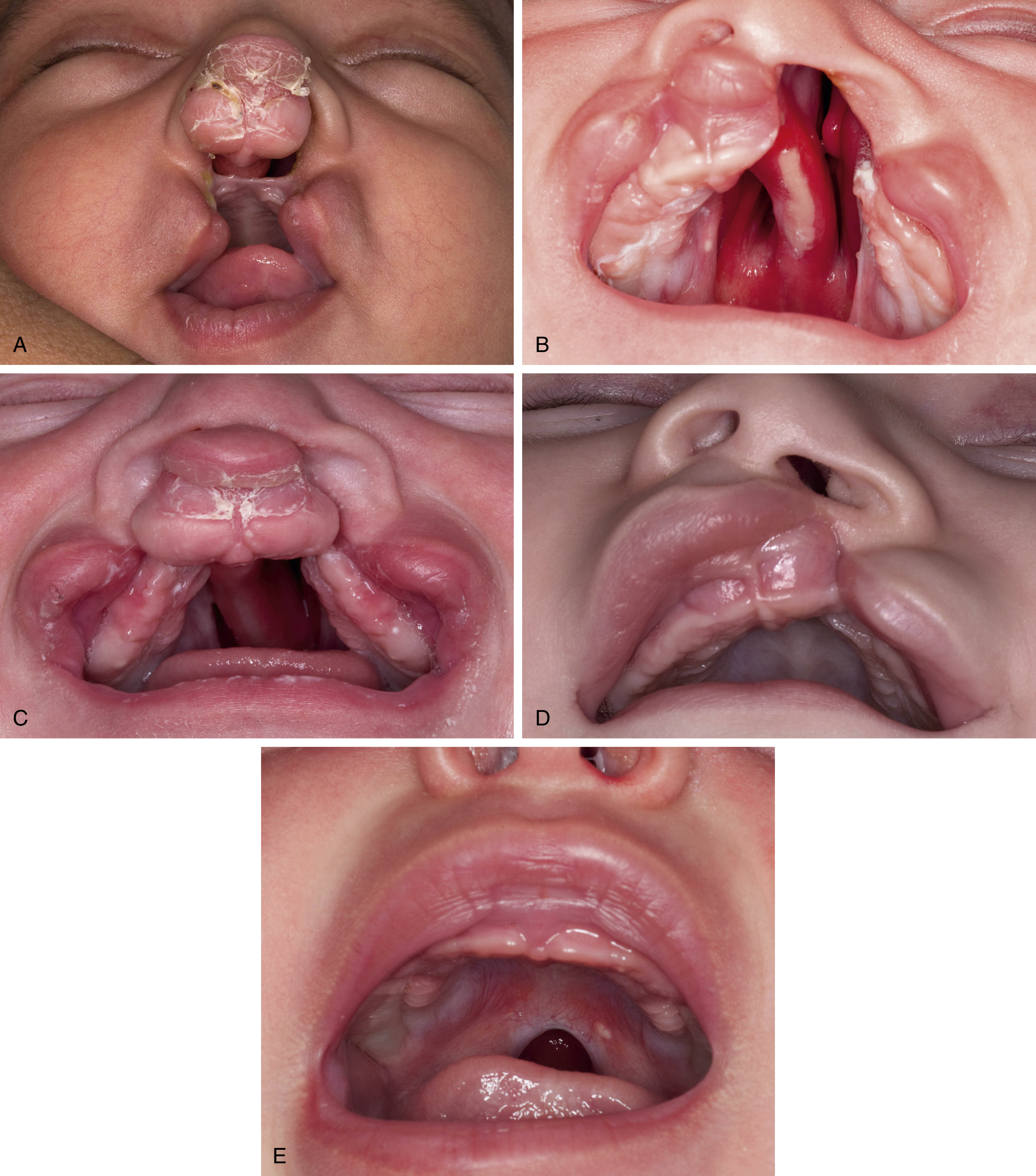
Isolated cleft palate is epidemiologically and etiologically distinct from cleft lip and palate. The incidence of cleft lip and palate varies by race, with approximately 2 in 1000 in Orientals, 1 in 1000 in Caucasians, and 0.4 in 1000 in Afro-Caribbean populations. Moreover, males are twice as likely to be affected than females. In contrast, isolated cleft palate occurs in 1 in 2000 live births across all ethnic backgrounds and more commonly in females, with a 2:1 predominance over males. Of all orofacial clefts, approximately 20% are isolated cleft lips, 35% isolated cleft palates, and 45% are clefts involving lip and palate. The etiology of clefting is multifactorial, including genetic factors, environmental factors and gene–environment interactions. Environmental risk factors include maternal obesity, diabetes mellitus, exposure to alcohol, smoke, diet, viral infections, drugs and various teratogenic agents, as well as physical and/or emotional stress. A strong association between the interferon regulatory factor 6 ( IRF6 ) gene and nonsyndromic cleft lip and palate has been demonstrated. It has been reported that aberrations of IRF6 comprise 12% of the genetic contribution to cleft lip and palate and increase the risk of recurrence threefold in families with one affected child. The familial association for cleft palate is commonly cited as follows:
- •
If normal parents have a child with a cleft palate, the frequency of cleft palate in the next child is estimated at 2%
- •
If normal parents with a family history of cleft palate have a child with cleft palate, the risk for the next child is 7%
- •
If normal parents have two children with cleft palate, the risk for the next child is 1%
- •
If one parent has a cleft palate and no previously affected children, the risk for the next child is approximately 6%
- •
If one parent has a cleft palate and a child with cleft palate, the risk for the next child is 15%.
Anatomy
The hard palate is composed of palatine processes of the maxilla and the palatine bone. The palate is supplied by the greater palatine vessels and nerves, which emerge through the greater palatine foramina in the posterolateral regions of the hard palate. The nasal mucosa of the hard palate is in continuity with the mucosa of the lateral nasal walls, and medially with the vomerine mucosa. The soft palate is the mobile palatal segment, with muscles that act synergistically to effect velopharyngeal closure. The sensory supply of the soft palate is derived primarily from the lesser palatine nerves, which are comprised of several branches that extend from the lesser palatine foramina in the palatal process of the sphenoid through the palatal aponeurosis.
From a functional perspective, the levator veli palatini is considered the most important muscle of the soft palate. This muscle originates from the petrous temporal bone and the junction of bony and cartilaginous portions of the Eustachian tube. It then courses inferiorly, anteriorly, and medially to enter the velum and join the levator from the opposite side, forming the levator sling . The levator is found in the middle third (40%) of the velum, between the two heads of the palatopharyngeus ( Fig. 17.2 ). The levator is innervated by the pharyngeal plexus (motor branches of cranial nerves IX, X, and XI). Moreover, it has been suggested that the muscle also receives innervation from cranial nerve VII. This is supported by findings of hemipalatal palsy in patients with hemifacial microsomia. The cumulative action of the levator is to pull the velum superiorly and posteriorly. In the cleft palate, the levator inserts into the cleft margin in the anterior half of the velum and not directly into the posterior edge of the hard palate , as is commonly cited.
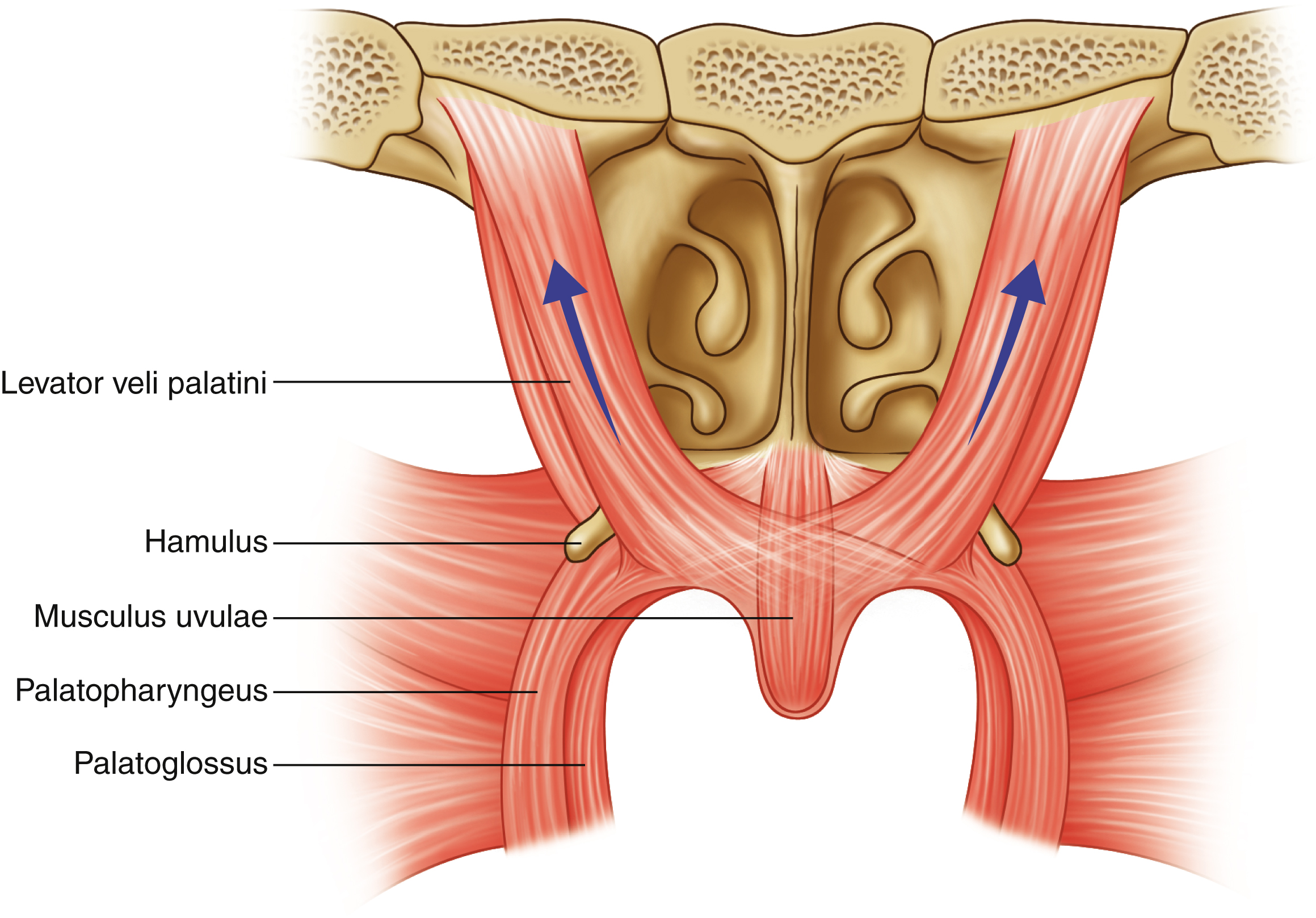
The palatopharyngeus originates from the palatine aponeurosis, and then divides into vertical and transverse fibers. The vertical fibers, palatothyroideus, form the posterior faucial (tonsillar) pillars, course in the lateral pharyngeal walls and insert into the greater horns of the thyroid cartilage of the larynx. The transverse component, palatopharyngeus proper, courses posteriorly along the lateral pharyngeal walls and intermingles with the superior pharyngeal constrictor muscle. The palatopharyngeus is also innervated by the pharyngeal plexus. Although most of the literature supports the notion that the palatopharyngeus is more active during swallowing than speech, the palatothyroideus fibres are in an anatomical position to act as an antagonist to the elevating action of the levator muscle but as a synergist in stretching the velum backwards. The palatopharyngeus proper assists in the medial displacement of the lateral pharyngeal walls during velopharyngeal closure and contributes to the formation of Passavant’s ridge . Further, muscular contraction of the palatopharyngeus stretches the velum inferiorly and laterally to increase the available velar surface area and optimize velar shape for contact with the posterior pharyngeal wall. In the midline of the cleft palate, the levator and palatopharyngeus fuse to form the “cleft muscle” as described by Veau. However, in contradistinction to the levator veli palatini, the palatopharyngeus also inserts into the posterior border of the hard palate.
The palatoglossus is the most superficial (oral) muscle of the soft palate, traverses through the anterior tonsillar pillars and inserts into the tongue. As such, contraction results in tongue elevation, palatal depression, and narrowing of the pharyngo-oral isthmus. The muscle does not have an ascribed role in velopharyngeal closure per se. The levator, palatopharyngeus, and palatoglossus diverge anteriorly to insert into the margins of the cleft, aberrant palatal aponeurosis, as well as the oral and nasal mucosa.
The superior pharyngeal constrictor is a quadrangular muscle, which originates from the medial pterygoid plates and pterygomandibular raphe. The muscle courses around the pharynx, forming the lateral and posterior pharyngeal walls, enclosing the nasopharynx and upper oropharynx. The muscle fibres diverge superiorly and inferiorly as they approach the posterior midline to insert into the pharyngeal ligament. The space between the superior margin of the muscle and cranial base is occupied in the lateral nasopharyngeal wall by levator and Eustachian tube and in the posterior pharyngeal wall by the pharyngobasilar fascia. The muscle, innervated by cranial nerve X, causes medial excursion of the lateral pharyngeal walls and, along with palatopharyngeus, anterior displacement of the posterior pharyngeal wall. The latter accounts for the formation of Passavant’s ridge. While historically considered a compensatory mechanism in patients with velopharyngeal insufficiency (VPI), much evidence exists to challenge this assertion. For instance, the ridge is often noted to be below and slower than palatal excursion.
The tensor veli palatini does not contribute to movement of the velum. The tensor originates from the scaphoid fossa of the greater wing of the sphenoid and membranous portion of the Eustachian tube. The muscle courses medially and inferiorly parallel to the levator and forms a tendon. The tendon hooks around the hamulus of the pterygoid plate (to which it is partially attached), assumes a horizontal orientation and inserts along the posterior border of the hard palate as the palatal aponeurosis, occupying the anterior third of the velum. In the cleft palate, the tensor palatini reaches the hamulus and appears to diverge into two components – a triangular tendinous insertion into the lateral posterior edge of the hard palate (does not form the palatal aponeurosis), in close proximity to the nasal mucosa, and a less substantial component, which passes toward the oral surface behind the greater palatine neurovascular bundle. Some authorities recognize a separate but closely related muscle, the dilator tubae, which rounds the middle third of the pterygoid hamulus without an insertion. There has been much debate as to whether the tensor (and/or dilator tubae), has a significant action on the velum and regarding the relative importance of the tensor (or dilator tubae) and levator in Eustachian tube function.
The musculus uvulae, thought to be supplied by the lesser palatine nerve, is a paired muscle located above the levator, attached to the palatal aponeurosis anteriorly and passing into the uvula posteriorly. The muscle is covered nasally and laterally by mucous glands. Contraction of the muscle may assist in velopharyngeal closure by adding to the bulk of the midline dorsal ridge over the levator eminence and also possibly by lifting the free border of the palate into deeper contact with the posterior pharyngeal wall. Others argue that this muscle may not be of importance in velopharyngeal function. In the cleft palate the presence of the musculus uvulae is controversial and rarely seen.
Finally, the salpingopharyngeus is a small muscle that originates from the torus tubarius and courses vertically along the lateral pharyngeal walls. While this muscle does not contribute to velopharyngeal closure, it may draw the lateral pharyngeal walls superiorly.
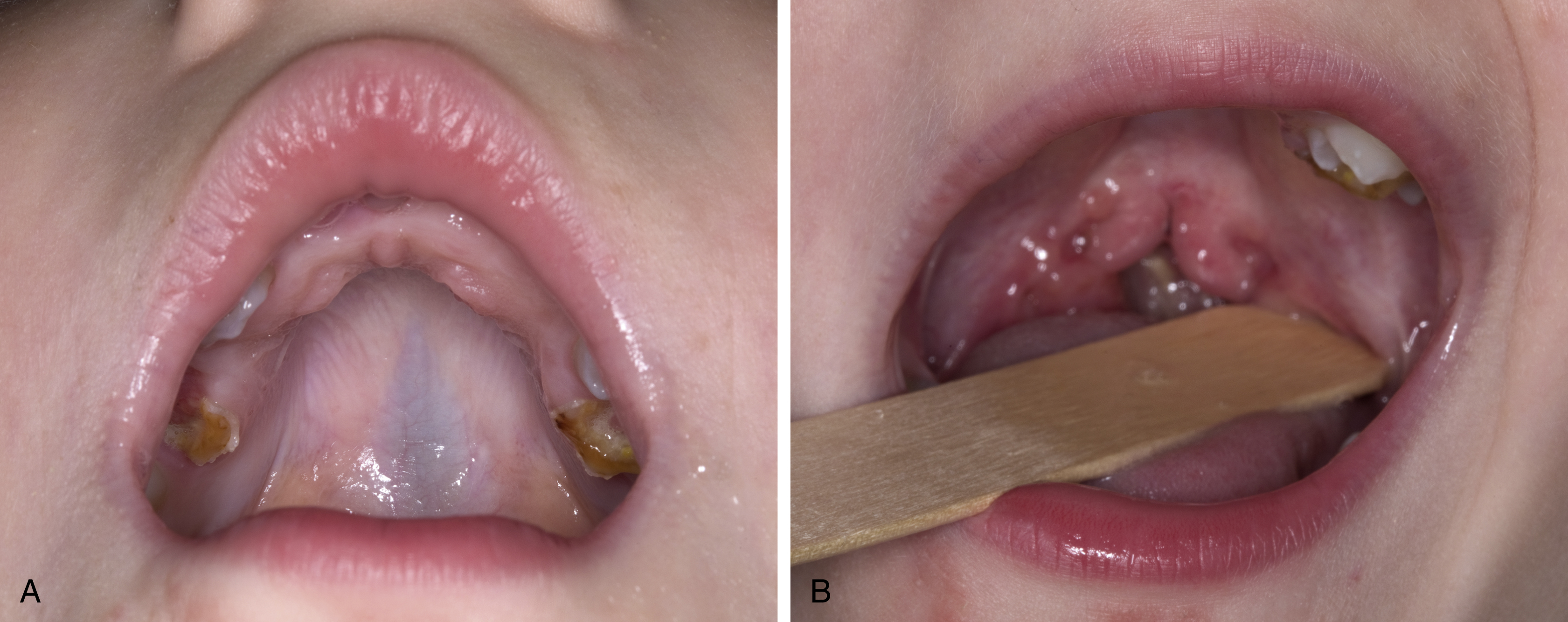
Submucous Cleft Palate
Submucous cleft palate is a form of cleft palate wherein the mucosa is intact but the underlying musculature is similarly abnormal to that of an overt cleft palate. In 1954, Calnan qualified the overt submucous cleft palate with a classic triad, including bifid uvula, palpable notch in the posterior hard palate, and zona pellucidum. The zona pellucidum is a midline translucent zone that represents diastasis of the velar musculature ( Fig. 17.3 ). Kaplan described the occult subcumous cleft palate, where velar malfunction is observed, but the aforementioned or typical features are not apparent. Given evidence of velopharyngeal dysfunction (VPD), the diagnosis of occult submucous cleft palate is often made following evaluation with nasopharyngoscopy, where a midline groove or concavity is seen on the nasal surface of the palate. The latter is thought to represent hypoplasia or agenesis of the musculus uvulae, as well as velar musculature diastasis. Sommerlad et al. argued that there was a spectrum of severity in submucous clefting and described a scoring system based on the severity of each feature of the triad. In effect, Kaplan postulated that isolated clefts of the secondary palate, overt, and occult submucous cleft palates are variations in expression of the same embryonic process. Diagnosis of submucous cleft palate is often delayed until speech development allows appreciation of VPD.
Associated Syndromes
While a syndrome occurs in approximately 10% of patients with cleft palate in association with cleft lip, associated syndromes are described in up to 50% of cases with isolated cleft palate.
Pierre Robin Sequence
The most commonly associated anomaly is Pierre Robin sequence, which occurs in approximately 1 in 8500–14,000 births. The condition typically occurs sporadically, but may be familial, with autosomal dominant mode of inheritance. The sequence includes micrognathia/retrognathia, glossoptosis, and respiratory distress. While not a qualification of the sequence, a cleft palate occurs in approximately 60%–90% of cases. Indeed, in his original description of 1923, Pierre Robin, the French stomatologist credited for the identification of these infants, failed to include cleft palate as an associated feature, but later added cleft palate as an aggravating factor. The cleft palate in the context of Robin sequence is typically U-shaped, as opposed to the classic V-shaped cleft palate, and implies a wider palatal gap. The theory for the occurrence of cleft palate in Robin sequence is that the posterior displacement of the mandible results in elevation and dorsiflexion of the tongue at approximately 9 weeks of gestation, preventing migration and fusion of the palatal shelves. The majority of cases (83%) present as a component of a named syndrome, including Stickler syndrome, velocardiofacial syndrome, Treacher Collins syndrome, fetal alcohol syndrome, Nager syndrome, oculo-auriculo-vertebral spectrum, Apert syndrome, ectrodactyly–ectodermal dysplasia clefting syndrome, Opitz-G/BBB syndrome, and orofacial digital syndrome. Thus, with the Robin sequence, the presence of other anomalous features should be considered ( Fig. 17.4 ).
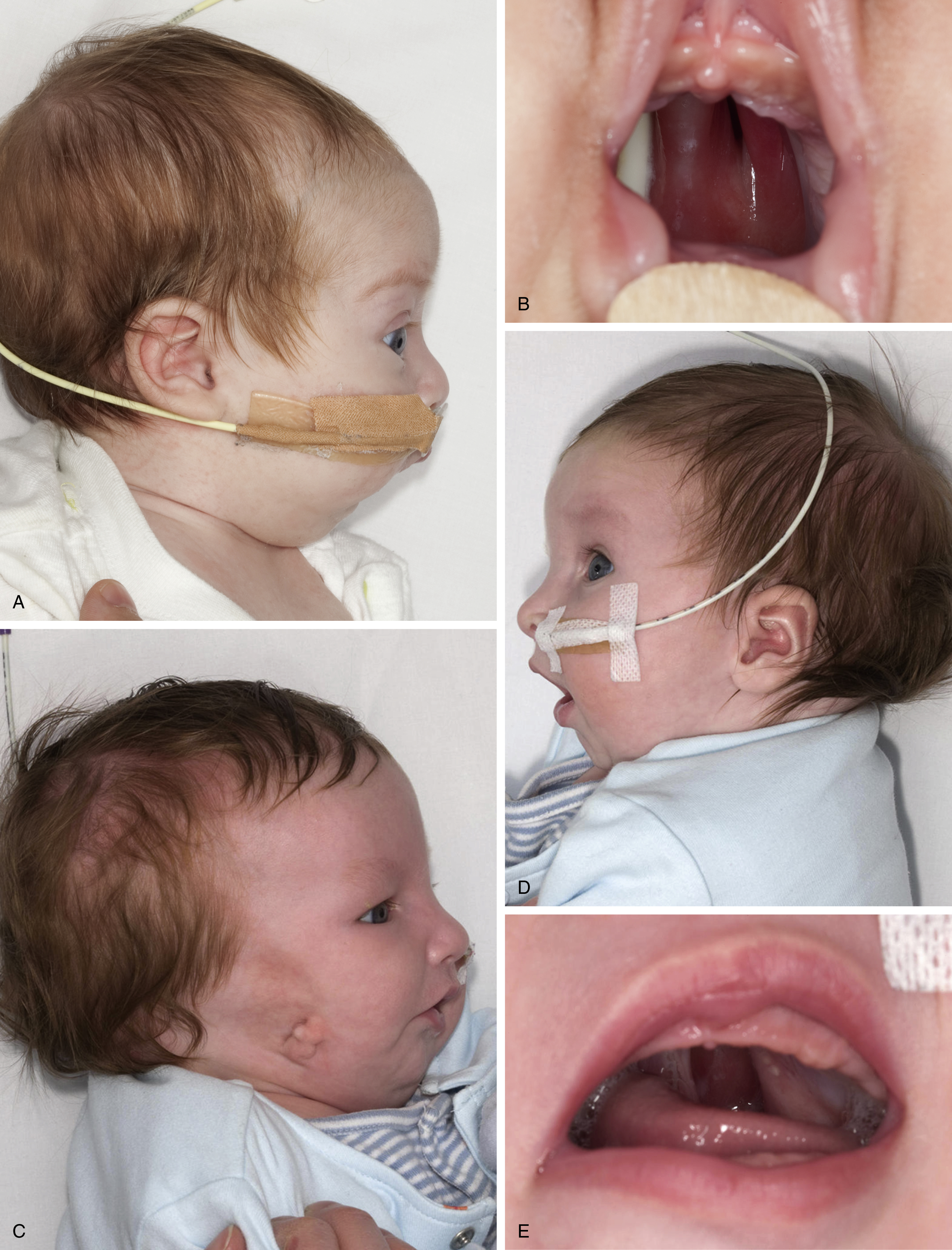
The glossoptosis in Robin sequence initiates a sequence of events including airway obstruction, increased energy expenditure, and impaired caloric intake from poor feeding. Thus, in the neonatal period, the primary objectives are to relieve airway obstruction and avert failure to thrive. However, glossoptosis is not the only contributor to upper airway obstruction in children presenting with Robin sequence. Acute angulation of the basicranium, pharyngeal, laryngeal and/or lingual anomalies, subglottic anomalies, hypotonia and central nervous system disorders may all cause or contribute to airway obstruction. Thus, microlaryngobronchoscopy is essential to determine the mechanism(s) of airway collapse and if obstruction can be relieved completely with treatment strategies that address tongue positioning alone. In effect, Sher et al, based on endoscopic observations, demonstrated four patterns of pharyngeal collapse in patients with craniofacial anomalies, any of which can be found in Robin sequence. In types 1 and 2, the retropositioned tongue is the primary etiologic factor responsible for obstruction, whereas in types 3 and 4 airway compromise is secondary to lateral pharyngeal wall collapse and pharyngeal stenosis, respectively. Thus, no single treatment will resolve respiratory obstruction in all patients, and management must be tailored to the specific underlying etiology. Airway compromise is assessed with oxygen saturation recordings and polysomnographic studies, the need for oxygen supplementation, and appropriate weight gain (with or without gavage feeding). Routine laboratory investigations and additional studies include serial blood gas analysis for elevated levels of carbon dioxide and serum bicarbonate, and pH recording for gastroesophageal reflux. As primary measures, management of airway compromise secondary to glossoptosis include prone or semiprone positioning and placement of a nasopharyngeal airway with or without continuous positive airway pressure. If the latter conservative treatment modalities fail to relieve airway obstruction, surgical intervention is warranted. Douglas popularized the technique of glossopexy, originally described by Shukowsky in 1911. This procedure was later modified by Routledge and various modifications were subsequently developed. The latter concept is based on the premise that airway obstruction is secondary to glossoptosis and surgical adhesion between tongue and lip can effectively mitigate the retro-displacement of the tongue. In 1983 a French stomatologist, Viviane Epois, asserted that the imbalance of the muscular insertion of the tongue on the mandible was responsible for the micrognathia, tongue elevation, and glossoptosis. More specifically, that the musculature of the floor of the mouth is under increased tension and impels the tongue superiorly and posteriorly, resulting in respiratory obstruction. The latter is the premise for the subperiosteal release of the floor of the mouth musculature for refractory cases of respiratory obstruction in the Robin anomalad. Following release, the tongue can adopt a more anterior position. Mandibular distraction is now a well recognized and widely used surgical alternative in patients with obstruction refractory to conservative measures. With this method the tongue is indirectly pulled forward through muscular attachment to the lingual surface of the mandible. In instances where the latter measures fail or in cases where other airway anomalies contribute to obstruction, tracheostomy is often required.
Stickler Syndrome
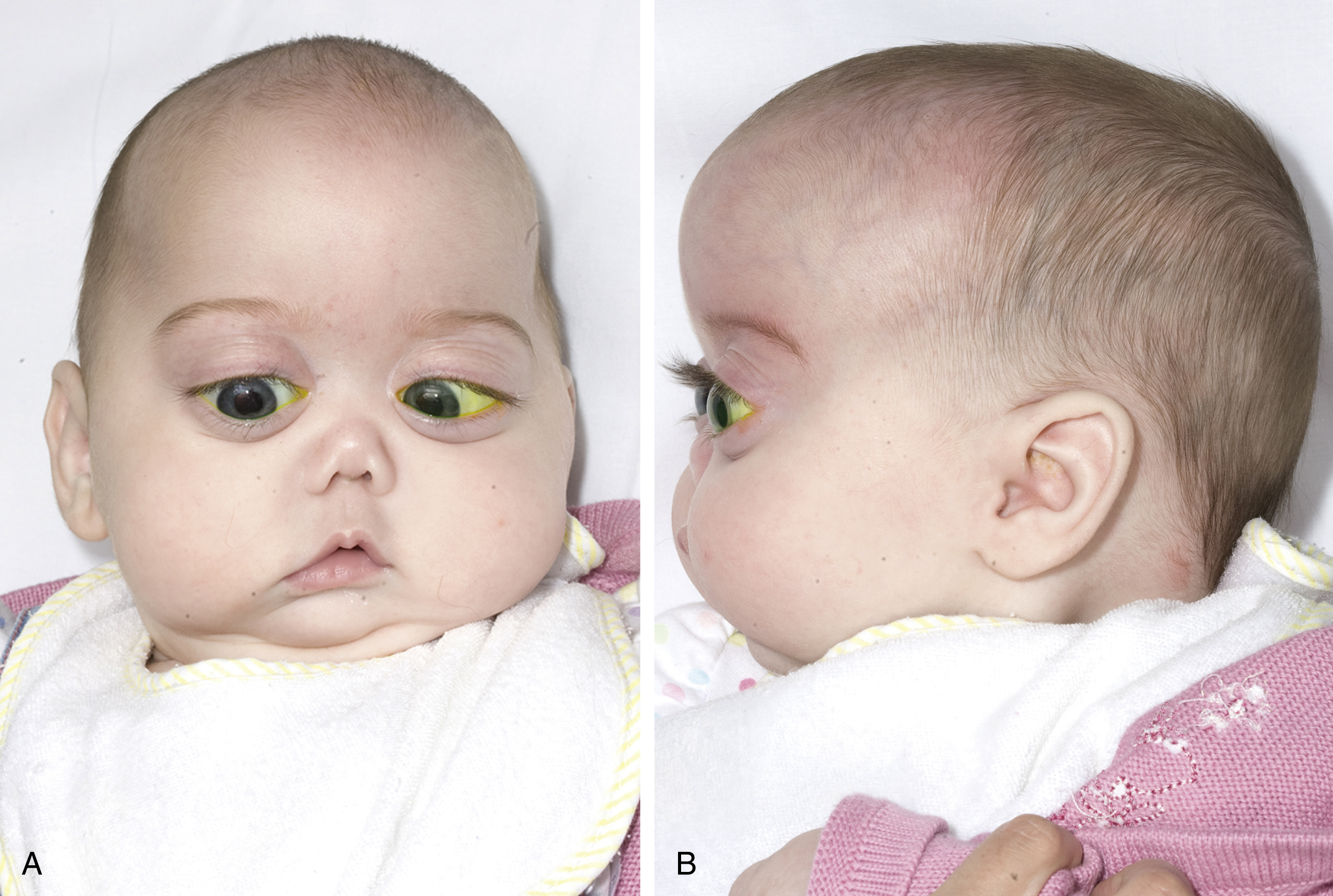
Stickler syndrome is a hereditary progressive arthro-ophthalmopathy with an estimated frequency of 1 in 10,000. It is an autosomal dominant connective tissue dysplasia characterized by cleft palate, degenerative joint changes, ocular manifestations, and progressive sensorineural hearing loss. Moreover, these patients often exhibit a characteristic flat facial appearance, secondary to hypoplasia of the midface and bridge of the nose ( Fig. 17.5 ).
Van der Woude Syndrome
While rare (1 in 100,000), van der Woude syndrome is an autosomal dominant syndrome with variable expressivity also associated with cleft palate. Van der Woude syndrome is associated with IRF6 mutations on chromosome 1q. Moreover, it is unique in that it may also be expressed as either cleft lip with or without cleft palate. A distinguishing feature of the syndrome is bilateral paramedian lower lip pits. IRF6 mutations are also responsible for the popliteal pterygium syndrome, which is characterized by cleft lip and/or palate, lower lip pits and additionally qualified by the presence of bilateral popliteal webs, hypodontia, syndactyly, nail abnormalities, anomalies of the genitourinary system, and ankyloblepharon. Interestingly, as previously mentioned, the IRF6 gene has been recognized to have the strongest genetic association with nonsyndromic cleft lip and palate. Indeed, some patients with van der Woude syndrome do not display the typical lower lip pits, and may be erroneously classified as nonsyndromic. Thus, IRF6 mutations should be considered in those with affected parent–child and especially segregating cleft palate and cleft lip and palate.
22q11.2 Deletion (Velocardiofacial) Syndrome
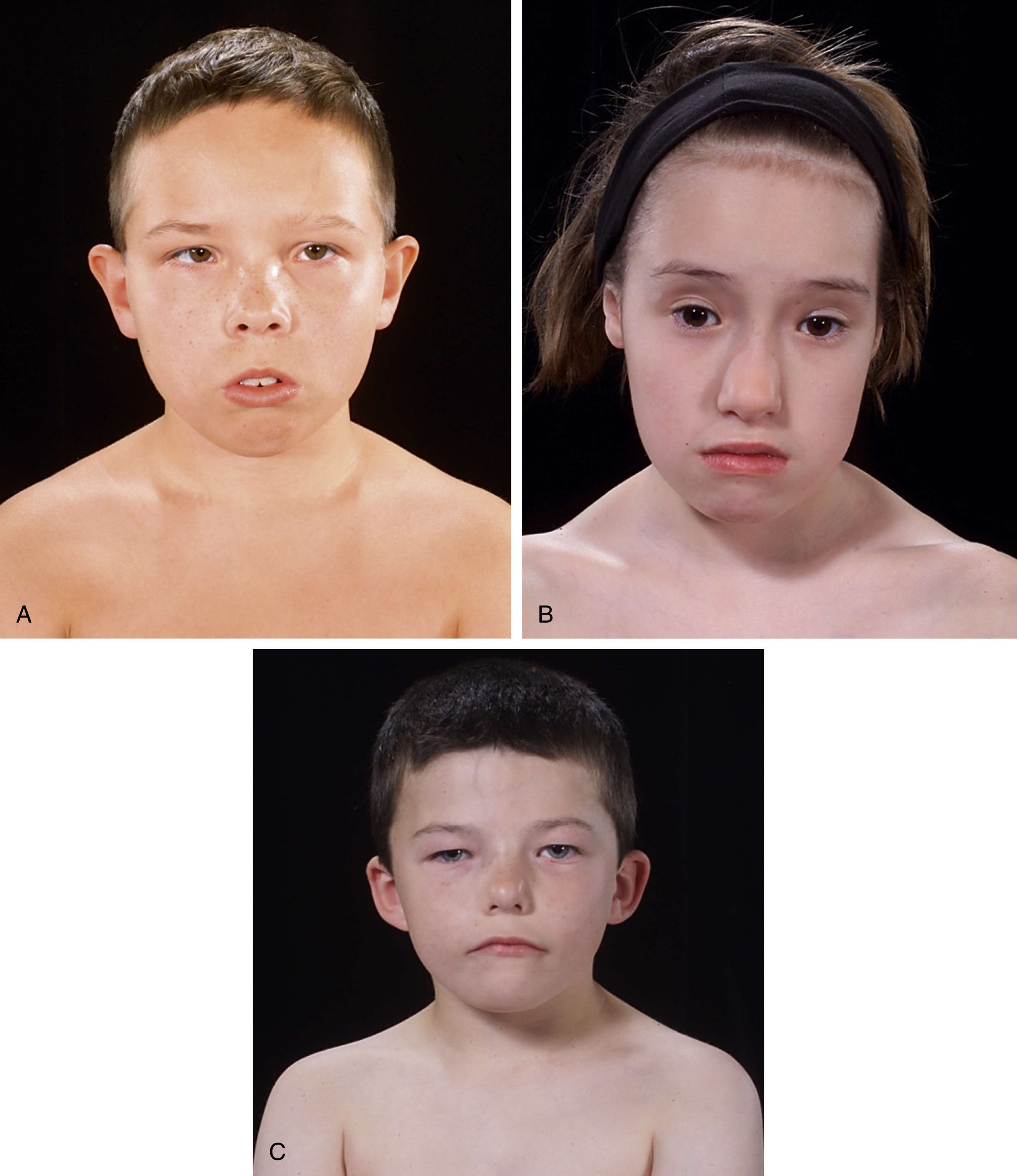
Although clinically under-recognized, velocardiofacial syndrome (VCFS) is the most common multiple anomaly syndrome in humans, with an estimated frequency of 1 in 2000–4000 live births and equal sex distribution. Microdeletions occur in the q11.2 region of chromosome 22, identified by fluorescence in situ hybridization (FISH) test. In these patients, the TBX1 gene is always deleted, with 90% of patients having a deletion of the typical deleted region (TDR; common 22q11.2 deletion, size 3 Mb), while 10% have a smaller deletion of only the proximal 1.5 Mb of the TDR (proximal deleted region, PDR). Both TDR and PDR include the TBX1 gene area. A few individuals with VCFS have shown unique 22q11.2 deletion intervals or a chromosome 10p14 deletion. In the majority of patients (>90%) 22q11.2 deletions occur as spontaneous events, with both parents unaffected. However, in approximately 10% of individuals, deletion is identified in a parent and thus there is autosomal dominant transmission with variable expression. There is phenotypic overlap between VCFS and DiGeorge, Sedlacková, conotruncal anomaly face and Cayler syndromes. Indeed, all these aforementioned syndromes are descriptions of the same entity, with over 180 separate anomalies or developmental sequences reported in association with VCFS covering nearly every organ and system. Notable among these are immune deficiency, hypocalcemia, conotruncal cardiac anomalies, short stature, learning difficulties, behavioral problems, psychiatric illnesses, and slender hands and digits. These patients also have a characteristic facial appearance, due to a combination of skeletal and soft tissue features. These include microcephaly, a long middle third of the face, flat zygomatic arches, mandibular retrognathia, broad nasal dorsum, narrow alar bases and bulbous nasal tip, hypotonic flaccid facies, narrow, downslanting palpebral fissures, and external ear anomalies ( Fig. 17.6 ). Also reported in patients with VCFS are aberrant carotid and subclavian arteries. Indeed, it has been hypothesized that the large number of vascular anomalies in major vessels reported in patients with VCFS may result in a number of developmental sequences. The latter notion is supported by reports of Robin, DiGeorge, Potter, and holoprosencephaly sequences in individuals with VCFS. The phenotypic expression in this syndrome is markedly variable with no clinical feature occurring in all cases and no reported case with all or even most of the associated clinical findings.
VCFS is now recognized as the most common syndrome associated with cleft palate and velopharyngeal insufficiency. The majority (approximately 75%) of children with VCFS have hypernasal speech and severe language impairment. Moreover, affected individuals commonly present with VPI, most often associated with overt or occult submucous cleft palate. While the frequency of VPI among individuals with nonsyndromic submucous cleft palate is approximately <9%, VCFS patients show VPI associated with submucous cleft palate in >70% of cases. In addition to overt or occult submucous cleft palate, there are several other factors that contribute to the high frequency of VPI in VCFS, including platybasia, congenitally short velum, palatopharyngeal disproportion, abnormalities of pharyngeal muscles with hypodynamic velopharynx, lymphoid tissue (adenoid) hypoplasia, and tonsillar hypertrophy. The latter anatomic and physiologic anomalies often render management of VPD in patients with VCFS a greater therapeutic challenge than in their non-syndromic counterparts, often necessitating a combination of palatoplasty and pharyngeal surgery. Despite the more extensive surgical correction required, speech outcomes are universally inferior when compared with those for non-syndromic patients.
Palatoplasty
Pathologic sequelae of cleft palate include feeding and nutritional difficulties, serous otitis media (with potential hearing loss), and abnormal speech development. Thus, the goals of cleft palate repair include improved feeding and normal speech, while minimizing the deleterious impact on maxillary growth and dental arch relationship.
In the neonatal period, feeding and adequate weight gain are a priority. Due to reduced ability to suck, and particularly to draw the nipple into the mouth, infants with cleft palate have difficulty feeding, and breastfeeding can only be achieved with considerable effort and modification of technique. In developed countries, most infants are bottle-fed, which allows a steady stream of milk, usually by a modified teat and soft bottle.
With regards to hearing, all patients with cleft palate should undergo a neonatal hearing assessment. Some argue that ventilation tubes should be inserted if there is a persistent conductive hearing loss of >45 dB on distraction testing or >35 dB on pure tone audiometry. However, given there is evidence that correction of the muscle abnormality in the cleft palate improves Eustachian tube function, others contend that insertion of ventilation tubes should be deferred.
Children with clefts generally do not have difficulties with swallowing and aspiration unless there is a coexisting intrinsic neuromuscular or anatomic abnormality of the tongue or pharynx. Evidence of the latter usually warrants further investigation with videofluoroscopy and endoscopy.
Timing of Repair
There is little debate that, in most situations, a cleft palate should be repaired. However, consensus is lacking on timing of repair. Restricted maxillary growth secondary to cleft palate repair has prompted some to promote two-stage palatoplasty, wherein the velum is repaired early in order to enable normal speech acquisition and hard palate closure delayed to facilitate maxillary development. Delayed closure of the hard palate may benefit facial growth but most reports suggest detrimental effects on speech. Irrespective, there is general agreement that soft palate repair should be effected before 12 months of age. The senior author aims to achieve complete palate closure by 6 months of age. There is evidence from a randomized controlled trial that speech outcome is superior following palatoplasty at 6 months as compared to 12 months. In this study, children with clefts repaired at 6 months of age did not develop compensatory articulations, while those with palate repair delayed to 12 months of age did display abnormal articulation patterns. Further, no difference in maxillary arch development, soft tissue profile or velopharyngeal dysfunction was noted between the two groups at 4 years from surgery. There is currently an international randomized controlled trial, the Timing of Primary Surgery for Cleft Palate (TOPS trial), comparing palate closure at 6 and 12 months of age.
For submucous cleft palate repair, timing is similar to overt cleft palate repair for infants with obvious evidence of a submucous cleft and persistent feeding difficulties. Where feeding is not a considerable issue, repair is otherwise indicated if an infant is not producing pressure consonants (/d/, /b/) by 15–18 months. In the older child with submucous cleft palate, surgical treatment is indicated only in the presence of consistent VPI or persistent hypernasality, as the presence of submucous cleft palate does not ensure VPD.
Preoperative Assessment
Appropriate preoperative assessment is mandatory to ensure that there is no history of airway compromise or anemia and there is adequate weight gain. The main contraindications to palate repair are associated medial comorbidities, which may present an unacceptable increased anesthetic risk and suboptimal weight.
Controversy exists regarding the benefits of palate repair in older children and adults. Arguably, adults with unrepaired clefts have compensatory patterns of articulation that are so well embedded that palate repair is unlikely to make much difference to speech. However, there are reports of some improvement in speech even though normality is not achieved. An international task force has produced a report and recommendations on delayed palate repair.
History of Palatoplasty
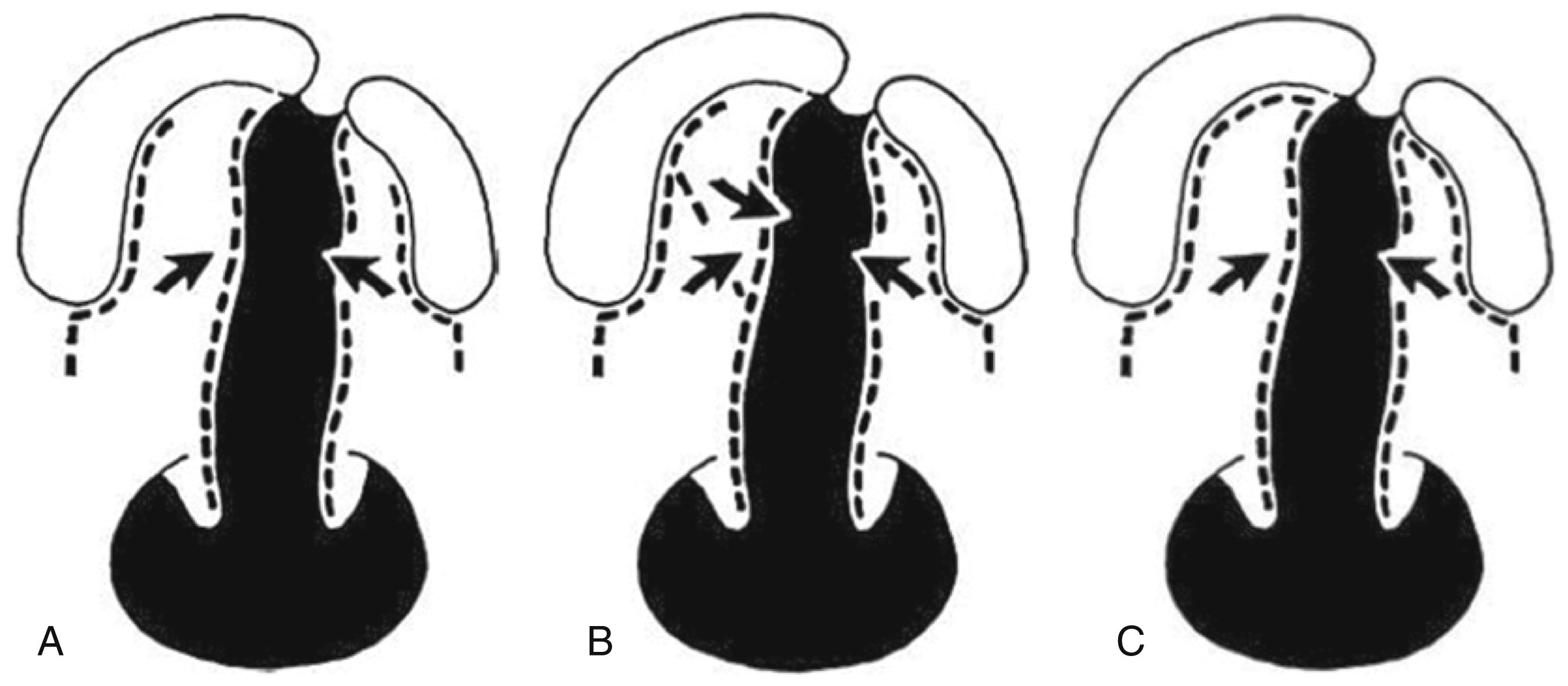
Before the development of general anesthesia, palate repair was performed on an awake patient. The first recorded repair was in France in 1819, when Roux repaired the palate of an Edinburgh medical student, John Stephenson, who went on to become professor of anatomy in McGill University, Montreal, Canada. In 1859, Bernhard von Langenbeck described bilateral bipedicled mucoperiosteal flaps for palate repair. Lateral relaxing incisions are made at the junction of gingival and oral palatal mucoperiosteum lateral to the greater palatine neurovascular bundle, in addition to cleft marginal incisions ( Fig. 17.7A ). Further developments in technique by Victor Veau, Thomas Kilner, and William Wardill were introduced to increase palatal length in an effort to aid in velopharyngeal closure ( Fig. 17.7B ). The three- or four-flap Veau–Wardill–Kilner pushback palatoplasty includes a V to Y incision and closure of the anterior hard palate. The resulting flaps are based on the greater palatine arteries posteriorly. Inherent in the pushback operation is release of the muscular attachments from the posterior edge of the hard palate. While the pushback palatoplasty lengthens the palate, theoretically retro-positioning the levator into a more favorable position, this has not been shown to be effective in improving speech. Moreover, by virtue of the fact that the bone is denuded anteriorly, this repair adversely affects maxillary growth and the single nasal mucosal layer anteriorly is associated with a higher rate of fistula. In effect, the latter procedure has been largely abandoned in modern era palatoplasty. The two-flap technique popularized by Janusz Bardach does not aim to achieve pushback, but is believed by its proponents to improve exposure of the underlying velar musculature ( Fig. 17.7C ). Many surgeons simply return the flaps to their original position to avoid leaving raw areas of bone. Arguably, this may not avoid impairment of maxillary growth.