Chemical Carcinogenesis: Introduction
|
Cutaneous Cancer and Public Health
According to the American Cancer Society, it is estimated that over 2,000,000 cases of basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), 46,770 cases of melanoma in situ, and 68,130 cases of invasive melanoma will be diagnosed that in the year 2010.1 The precise number of nonmelanoma skin cancers is unknown since they are not routinely reported to registries, but recent data suggest that previous approximations are likely to have been underestimates.2 Although only a small fraction of patients with nonmelanoma skin cancer will die as a result of their cancer, which is SCC in nearly all cases, the frequency of these cancers nonetheless results in an estimated 1,000 and 2,000 deaths per year. In contrast, although much less common than nonmelanoma skin cancer, melanoma has a continually rising death rate now estimated at 8,700 per year, and it is currently predicted that the lifetime risk in Caucasians of developing melanoma is a staggering 1 in 37 for males and 1 in 56 for females.1 Because they are so common, cutaneous cancers have a major impact on health care costs: in addition to the mortality burden, treatment is associated with considerable morbidity and cosmetic defects. For these reasons, understanding the etiology and pathogenesis of these malignancies is a significant public health goal, and development of rational nondeforming therapies to reduce morbidity and mortality is urgently needed. The high prevalence of skin cancer, the external location of the tumors, and well-defined preneoplastic lesions all provide an excellent opportunity for studying the factors regulating cutaneous cancer induction in humans. Those qualities that facilitate the study of cutaneous neoplasms in human populations have also been useful in establishing relevant animal models. Advances in molecular genetics, keratinocyte cell culture, and development of genetically altered mice and reconstructed human skin models have greatly facilitated the analysis of basic mechanisms of cutaneous carcinogenesis. Our main focus in this chapter will be on nonmelanoma skin cancer: the reader is referred to Chapter 124 for further discussion of melanoma.
Agents Associated with Skin Cancer Induction in Humans
The ultraviolet radiation (UVR) in sunlight is the primary etiologic agent for all skin cancers, and thus UVR is the major carcinogen in the human environment. The powerful carcinogenic activity of UVR is attributable to its ability to damage DNA and cause mutations, its capacity to clonally expand incipient neoplastic cells whose altered signaling pathways provide a survival advantage in the face of ultraviolet-induced cytotoxicity, its ability to induce reactive oxygen species, and its activity as an immune suppressant (see Chapter 112). The association of UVR with skin cancer is so strongly supported by clinical, epidemiologic, and experimental data that it represents the most clear-cut etiologic factor in human malignancy.
Also implicated in the development of human skin cancer are various chemicals, as a result of environmental, occupational, or medicinal exposures (Table 111-1). In 1775, Sir Percivall Pott3 attributed the increased incidence of scrotal cancer in chimney sweeps to repeated exposure to soot. This report provided the first link between occupational exposure and the development of cancer as well as the first example of chemical carcinogenesis. According to the National Toxicology Program’s 11th Report on Carcinogens, published in 2005 (http://proxy.library.upenn.edu:6378/ntp/roc/toc11.html), 246 agents are listed as known or likely human carcinogens, and the great majority of these are chemicals. In the last 35 years, the mechanisms by which chemicals cause cancer have been unraveled, and reveal striking similarities to the properties responsible for UVR carcinogenicity, namely DNA damage, selective cytotoxicity, and immune suppression.
Agent | Individual at Risk | Route of Exposure | Tumor Types |
|---|---|---|---|
UV radiation | General population | T | BCC, BD, SCC, M |
Cigarette smoke | Smoker | T or S | SCC |
Soot | Chimney sweep | T | SCC |
Coat tar, pitch | Coker of coal, steel worker | T | SCC |
Petroleum oils | Machinist, textile worker | T and S | SCC |
Arsenic | Agriculture worker, living in areas of high groundwater and soil contamination | S and/or T | BD, SCC, BCC |
4,4′-Bipridyl | Pesticide manufacturer | T | SCC, BD |
Polychlorinated biphenyl | Petrochemical worker | T or S | M |
Dry cleaning reagents | Dry cleaner | T or S | BCC |
Fiberglass | Insulator | T | BCC |
Psoralen (PUVA) | Psoriasis patient | T and S | SCC, BCC, M |
Nitrogen mustard | CTCL patient | T | SCC |
Immunosuppressants | Transplant recipient, etc. | S | SCC, BCC |
Ionizing radiation | Cancer therapy patient | T | BCC, SCC |
The carcinogenic potential of coal and petroleum derivatives is now firmly established as a result of experimental animal studies and epidemiologic reports. Petroleum products, grease as well as insecticides, herbicides, and fungicides are particularly pathogenic for SCC, while fiberglass and dry-cleaning agents increase the incidence of BCC.4 Cigarette and pipe smokers have an overall twofold increased risk for cutaneous SCC, and the risk increases with the intensity of the tobacco use.5 Arsenic exposure is associated with the development of premalignant keratoses, Bowen’s disease, SCC, and BCC, as well as a number of internal malignancies.6 While Fowler’s solution (1% potassium arsenite) is no longer used in medical practice, certain herbal medicines are still a source of arsenic exposure. Occupational exposures to arsenic as a component of agricultural pesticides, sheep and cattle dip, mining and smelting, glass manufacturing, and other industries are well documented. A more insidious source of environmental arsenic exposure is contaminated drinking water or shellfish, and analysis of affected populations shows a dose-dependent increase in skin cancer.7 Mouse models have provided evidence for an interaction of ingested arsenic acting as a cocarcinogen with solar radiation to increase the frequency and size of cutaneous carcinomas and reduce the latency period.8,9
A variety of medications have been associated with the development of skin cancer. Systemic treatment with immunosuppressive agents results in an increased incidence of both benign and malignant skin lesions,10 which is generally attributed to reduced immune-surveillance of nascent tumor cells. However, a more specific mechanism predisposing to skin cancer has been proposed for azathioprine, which may sensitize DNA to ultraviolet A (UVA) radiation via its metabolite 6-thioguanine, leading to the production of mutagenic reactive oxygen species.11 In addition, cyclosporine A inhibits DNA repair and cell death in UVB-irradiated keratinocytes,12 raising the possibility that this compound may also increase photocarcinogenesis. Topical nitrogen mustard also increases the risk of developing skin cancer.13 Despite the clear-cut association between occupational exposure to tars and skin cancer, the use of coal tar in patients with psoriasis or eczema does not appear to increase the risk of developing skin cancer, based on a large study including over 13,000 patients.14 In contrast, the use of ionizing radiation to treat a variety of skin diseases increases the risk of BCC in all treated patients, and SCC in individuals with sun-sensitive skin.15
Systemic administration of 8-methoxypsoralen combined with UVA treatments (PUVA) is associated with a dose-dependent increase in the risk of developing cutaneous SCC that persists following discontinuation of therapy.16 Patients receiving high-level exposures (≥337 PUVA treatments) had over a 100-fold increase in SCC incidence while those receiving <100 treatments had a fivefold higher than expected incidence. 3.8% of individuals with PUVA-associated SCC developed metastases, and SCCs developed on the male genitalia much more frequently than in the general population.17 The incidence of BCC and melanoma is also increased in PUVA patients.18,19 PUVA is thus a potent stimulus for the induction of epithelial skin cancer in humans and also induces SCC in mice.20 While this implies that PUVA is a complete carcinogen by virtue of its DNA damaging properties, the frequent detection of human papilloma virus DNA in PUVA-induced cancers suggests that immunosuppression may also contribute to carcinogenesis in this setting.21 In contrast to PUVA, UVB phototherapy does not appear to be associated with an increased risk of developing nonmelanoma skin cancer.22 There is strong evidence implicating the use of tanning beds in the development of both nonmelanoma skin cancers and melanoma.23 The World Health Organization now places tanning beds in the category of group I carcinogens,24 which will hopefully help bring about policy changes leading to strict regulation of the tanning bed industry.25
The Nature of Chemical Carcinogens: Chemistry and Metabolism
How can such a diverse group of chemical and physical agents contribute to cutaneous cancer when the great majority of environmental agents to which humans are exposed are not carcinogenic? We now know that carcinogens can be genotoxic, nongenotoxic, or both.26 Genotoxic carcinogens have high chemical reactivity (such as alkylating agents like nitrogen mustard) or can be metabolized to reactive intermediates by the host (such as petroleum products). They form covalent adducts with macromolecules and target DNA in the nucleus and mitochondria.27 Since there is a good correlation between the ability to form covalent DNA adducts and the potency to induce tumors in laboratory animals, DNA is considered the ultimate target for most genotoxic carcinogens. The interaction with DNA is not random, and each class of agents reacts selectively with purine and pyrimidine targets.27 Furthermore, targeting of carcinogens to particular sites in DNA is determined by nucleotide sequence, by host cell, and by selective DNA repair processes making some genetic material at risk over others. As expected from this chemistry, genotoxic carcinogens are potent mutagens, particularly adept at causing base mispairing or small deletions, leading to missense or nonsense mutations. Others may cause macrogenetic damage such as chromosome breaks and large deletions. In all cases, mutations detected in tumors represent a combination of the effect of the mutagenic change on the function of the protein product and the effect of the functional alteration on the behavior of the specific host cell type.
A number of chemicals that cause cancers in laboratory rodents and contribute to human skin cancer incidence are not demonstrably genotoxic.28 Synthetic pesticides and herbicides, dry-cleaning reagents, and arsenic may fall within this group. The mechanism of action by nongenotoxic carcinogens is controversial and may be related in some cases to toxic cell death and regenerative hyperplasia. Induction of endogenous mutagenic mechanisms such as DNA oxyradical damage,29 depurination of DNA, and deamination of 5-methylcytosine may contribute to carcinogenicity of these agents. Nongenotoxic carcinogens may interfere with host protective mechanisms as has been suggested for the action of arsenic in suppressing DNA repair or inhibiting the activity of tumor suppressor genes.8 Thus, nongenotoxic carcinogens may serve as modifiers in concert with genotoxic agents such as UVR.
While a number of carcinogens can directly interact with DNA, many require bioactivation by cellular metabolic enzymes to form a compound that can react and form adducts with DNA. In general, these reactive intermediates are inadvertent byproducts of xenobiotic detoxification pathways. These pathways are complex and interactive,30 and genetic polymorphisms both in animal models and humans contribute to cancer susceptibility.31 This genetic variation in metabolic enzymes leading to differing rates of biotransformation and detoxification among individuals provides an approach to estimate individual risk profiles for particular chemical exposures. Initial stages in metabolic activation are most often carried out by cytochrome P450s, a class of heme containing monooxygenases, although other enzymes can be involved.32,33 Oxidation of carcinogens at carbon–carbon double bonds or saturated carbon atoms, or oxidation or reduction at nitrogen moieties, yields reactive intermediates that can be further metabolized and detoxified or can bind to DNA. Further biotransformation involves enzymatic conjugation of one of several different chemical groups such as glucuronide, glutathione, acetyl, or sulfate to reactive intermediates to enhance elimination. However, these conjugation reactions can also activate as well as detoxify carcinogens.30 A number of metabolic pathways and enzymes such as cytochrome P450s are induced by carcinogen or other chemical exposures through interaction of these xenobiotics with the aryl hydrocarbon receptor (AhR) a cytoplasmic receptor that functions as a transcription factor for detoxifying enzymes.34 Thus, carcinogen exposure induces detoxifying enzymes, which in turn increase cancer risk through biotransformation. Expression of these metabolic pathways can be modified by diet and hormones, thus, adding further complexity to the process and determination of relative risk of carcinogenesis.
Oncogenes, Tumor Suppressor Genes, and Hereditary Cancer Syndromes
If DNA is the target for carcinogens and mutagenesis is the underlying mechanism of cancer pathogenesis, how do we identify the genes that when mutated are responsible for tumor formation? Studies on the molecular basis of cancer development in the last 30 years have revealed two classes of genes, oncogenes and tumor suppressor genes that play a key role in the pathogenesis of cancer. An oncogene is any gene that can transform normal cells in culture and induce cancer in animals. Most oncogenes are derived from proto-oncogenes: normal cellular genes that are critical positive regulators of cell proliferation or inhibitors of apoptosis. Conversion to an oncogene can occur through point mutations resulting in a constitutively active protein, through DNA amplification, or through chromosomal translocations that link a highly active promoter with the proto-oncogene. Both of these later two mechanisms cause increased or inappropriate expression of a proto-oncogene and altered growth regulation of the normal cell. Tumor-suppressor genes normally function to negatively regulate cell proliferation, cause apoptosis, repair damaged DNA, or induce cellular differentiation. In contrast to oncogenes, both copies of a tumor suppressor gene must be inactivated to promote tumor development. Frequently, inactivating point mutations occur in one copy of a tumor suppressor gene, and the remaining normal copy is lost through a process of chromosomal missegregation during mitosis that leads to loss of heterozygosity.35,36
Which oncogenes and tumor suppressors contribute to development of cutaneous neoplasms? Considerable insight into the genetic basis of sporadic skin cancers has come from the elucidation of specific genes or genetic loci that define hereditary skin tumor syndromes (Table 111-2).37 In particular, the importance of DNA as a target for carcinogenesis was strongly supported by the discovery of defects in DNA repair genes that comprise the complementation groups of skin cancer prone xeroderma pigmentosum (XP) patients (Chapters 110 and 139).38,39 At least six independent genes, on distinct chromosomal loci, define proteins involved in nucleotide excision repair, a process critical for the protection of DNA against both UVR and chemical carcinogen-induced damage.40 Among these are proteins that recognize and bind to sites of DNA damage (XPA, XPC, XPE), helicases (XPB, XPD) and endonuclease components (XPG, XPF, defects in any of which give a skin cancer-prone phenotype. Potential polymorphisms with functional consequences in these and other DNA repair genes may contribute to susceptibility states in the general population as well.41–43 Examination of SCCs and BCCs from XP patients has also revealed potential target genes for cancer induction since signature mutations in PTCH1, RAS, p53, and INK4a-ARF (p16INK4a and ARF) have been found with high frequency.44
Gene | Type | Function | Locus | Tumor Type | Syndrome | Spontaneous |
|---|---|---|---|---|---|---|
TP53 | Tumor suppressor | DNA repair, apoptosis, cell cycle regulation | 17p13.1 | BCC, SCC | Li Fraumeni (but no increase in skin cancers) | Yes |
XPA, XPB XPC, XPD XPF, XPG PolH (XPV ) | Tumor suppressor | DNA repair | 9q22.3, 2q21 3p25, 19q13.2-q13.33 16p13.3-p13.13, 13q33 6p21.1 | BCC, SCC, melanoma | Xeroderma pigmentosum | Possible |
PTCH1 | Tumor suppressor | Hedgehog receptor | 9q22.3 | BCC, trichoepithelioma | Nevoid basal cell carcinoma | Yes |
SMO | Oncogene | Hedgehog effector | 7q31-q32 | BCC | Unknown | Yes |
p16(INK4A) | Tumor suppressor | Cyclin-dependent kinase inhibitor | 9p21 | Melanoma, SCC | Familial melanoma | Yes |
BRAF | Oncogene | Cell cycle, survival | 7q34 | Melanoma | None | yes |
HRAS, NRAS | Oncogene | Cell cycle, survival | 11p15.5, 1p13.2 | SCC, melanoma | None | Yes |
CTNNB1 | Oncogene | Cell–cell adhesion, transcription factor | 3p22-p21.3 | Pilomatricoma | Unknown | Yes |
LEF1 | Oncogene | Transcription factor | 4q23-q25 | Sebaceous tumors | Unknown | Yes |
CYLD | Tumor suppressor | NF-κB inhibitor | 16q12-q13 | cylindroma | Multiple cylindroma, multiple trichoepithelioma | Yes |
PTEN | Tumor suppressor | Phosphatase | 10q23.3 | Trichilemmoma melanoma | Cowden’s | Unknown |
MSH2 MLH1 | Tumor suppressor | Mismatch repair | 2p22-p21 3p21.3 | Sebaceous gland carcinoma | Muir–Torre | Unknown |
FLCN | Tumor suppressor | Ribosomal protein S6 modulator | 17p11.2 | Fibrofolliculoma | Birt–Hogg–Dube | Unknown |
Unknown | Unknown | 9p21 | Trichoepithelioma | Multiple trichoepithelioma | Unknown | |
Unknown | Unknown | Xq24-q27 | BCC | Bazex | Unknown | |
Unknown | Unknown | 9q31 | Keratoacanthoma | Ferguson-Smith | Unknown |
Chromosomal mapping studies in the basal cell nevus syndrome (Chapter 116), coupled with genetic and functional studies in Drosophila and mice, implicated mutations in the PTCH1 gene and other defects in the Hedgehog (Hh) signaling pathway in the development of hereditary and sporadic BCCs45 and a variety of internal malignancies.46 All of these tumors are characterized by elevated Hh signaling activity (Chapter 115). The INK4a-ARF locus, specifically mutations in the p16(INK4A) gene, was identified in the etiology of melanoma47 through mapping of the inheritance pattern of familial melanoma. Oncogenic mutations in BRAF were discovered in a high proportion of melanoma by systematically screening for alterations in genes in the Ras/MAP kinase signaling pathway,48 an important growth regulator in many cell types. Detection of specific mutations in Cowden syndrome (PTEN),49,50 Muir–Torre syndrome (MSH2, MLH1),50,51 multiple cylindroma (CYLD1),52 multiple trichoepithelioma (CYLD1),53 Birt–Hogg–Dube syndrome (FLCN),54 and sporadic pilomatricomas (CTNNB)55 and sebaceous tumors (LEF1)56 have illuminated genetic defects associated with the development of adnexal tumors. The delineation of the specific genes mutated in other syndromes where locus mapping is confirmed (Table 111-2) should provide even more insight into the molecular pathogenesis of a broader spectrum of skin neoplasms.
Animal Models Verify the Genetic Basis for Cutaneous Neoplasms
Using human hereditary cancer syndromes as a guide, genetically based animal models have been developed that validate the contribution of individual genes to human cancer susceptibility by prospective experimental analysis.57 These models have also contributed to the understanding of gene–environment interactions by testing the influence of chemical and physical carcinogens on tumor development in the setting of an altered genetic makeup. The discovery of inactivating PTCH1 mutations and activating SMO mutations in BCCs (see Chapters 115 and 116) led to the development of several mouse models exploring the role of deregulated Hh signaling in BCC tumorigenesis.45 Keratinocyte-targeted overexpression of SHH58,59 or an activated form of the Hh pathway signaling molecule SMO (M2SMO)60,61 resulted in the development of basal cell-like proliferations or follicular hamartomas in mouse skin. Overexpression of SHH in human keratinocytes grafted onto immune-deficient mice also resulted in BCC-like lesions.62 Mice with heterozygous disruption of the Ptch1 gene have features in common with basal cell nevus syndrome patients,63,64 including microscopic hair follicle-derived tumors and various macroscopic skin tumors, including BCCs, after treatment with UVR or ionizing radiation.65,66 When GLI1 or Gli2 transcription factors, which are nuclear effectors of Hh signaling are targeted to mouse keratinocytes, multiple skin tumors develop. GLI1 mice display a variety of follicle-derived tumors with relatively few BCCs,67 while Gli2 mice develop BCCs exclusively.68 Interestingly, when Gli2 expression was extinguished in gene-switch mice, BCCs regress but leave behind a subset of residual tumor cells, at least some of which may be able to form growing tumors when the Hh pathway is reactivated.69 Collectively, these findings provide strong evidence supporting the notion that constitutive Hh signaling plays a key role in, and may be sufficient for, BCC development.
The multistage induction of cutaneous SCC (Fig. 111-1)70 and the absence of a hereditary syndrome that specifically imparts uniquely high susceptibility to this lesion have made the delineation of specific genetic loci involved in SCC induction more complex. Nevertheless, the analysis of genetic defects in human SCCs has focused attention on specific targets such as DNA repair genes, RAS, p53, p16(INK4A), and the epidermal growth factor receptor (EGFR) pathway. Significant progress has been made in defining pathways capable of driving nonmelanoma skin cancer development using reconstructed human skin, comprising genetically altered primary human keratinocytes on architecturally intact dermis, grafted onto immune-deficient mice.71 Using this powerful system for modeling human skin cancer in mice, it has been shown that coexpression of oncogenic Ras and either Cyclin-dependent kinase 4 (Cdk4), or the NF-κB inhibitor IκB, in human keratinocytes, rapidly leads to invasive SCC development in reconstructed skin grafts.72,73
Figure 111-1
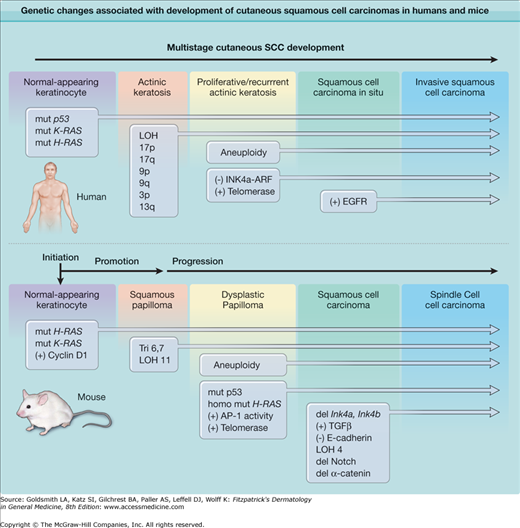
Genetic changes associated with development of cutaneous squamous cell carcinomas (SCC) in humans and mice. The multistage evolution of invasive squamous cell cancer in humans is depicted schematically with frequently associated genetic changes. Single base mutations in early lesions frequently are characteristic of ultraviolet light-induced damage, while later changes are associated with genomic instability. Increased activity of telomerase (deletion of inhibitor) or epidermal growth factor receptor (EGFR) tyrosine kinase (gene amplification) may also result from epigenetic changes. In chemically induced mouse cutaneous SCC, the multistage evolution to spindle cell tumors in this model is highly ordered both temporally and genetically. Operationally defined stages include initiation, promotion, and progression. Ras mutations are characteristic of chemical mutagens used to initiate tumor formation. Early upregulation of Cyclin D1 and later upregulation of TGFβ1 occur through epigenetic mechanisms and appear to be important components of carcinogenesis. Note that while p53 alterations are seen at early stages of human SCC development and ultraviolet radiation (UVR)-induced mouse SCC development (not shown), p53 mutations occur at later stages during mouse chemical carcinogenesis. (Modified from Dlugosz A, Merlino G, Yuspa SH: Progress in cutaneous cancer research. J Invest Dermatol Symp Proc 7:17, 2002, with permission of Nature Publishing Group, London, UK.)
Mouse models for XPA, XPC, and XPE deficiency have been constructed, and all show sensitivity to UVR and enhanced development of SCC after either UVR or chemical carcinogen exposure.39 A number of other DNA repair-deficient mutant mice are also being tested for cancer susceptibility and may reveal the mechanistic basis for new clinical syndromes by reverse genetics.74 Mouse mutants targeting the Ras gene have revealed important information concerning the contribution of the Ras pathway to particular stages of SCC development. Heterozygous activating Ras gene mutations are sufficient to induce a benign squamous papilloma, the precursor to SCC, and the yield of chemically induced tumors in mice genetically deleted in the H-ras allele is markedly reduced.75 Homozygosity of a mutant ras gene is associated with progression to SCC,76 suggesting that high penetrance of the Ras pathway can recruit additional changes required for progression. Transgenic targeting of oncogenic Ras to suprabasal epidermis produces terminally benign tumors, while mosaic targeting to basal cells or hair follicle outer root sheath cells encourages progression to SCC,77 indicating the target cell for early mutations may also determine the tumor phenotype.78 While heterozygous p53 inactivation is detected early in human SCC, p53 deletion in mice enhances malignant progression but does not increase benign tumor formation.79 This may be analogous to the common loss of heterozygosity seen at the p53 locus later in human SCC development.80 Of particular interest for human tumor development is the frequent observation that double heterozygotes for p53 deletion and other cancer-prone phenotypes (such as DNA repair deficiency) further sensitize mouse skin to tumor induction by UVR or chemical carcinogens. Mice with genetically defined defects in the p16Ink4a locus or its downstream target Cdk4 are sensitive to both squamous tumor and melanoma induction after treatment with carcinogenic chemicals, consistent with defects in this pathway detected in both melanoma and nonmelanoma human skin cancers.81,82 Elegant mouse modeling studies have confirmed the importance of Braf mutations, when combined with alterations in the tumor suppressor Pten, in the development of metastatic melanoma in mice.83
Tumor Biology and Biochemistry
Malignant tumors exhibit fundamental alterations in behavior that distinguish them from the normal tissues in which they arise. These differences include a reduced requirement for growth stimuli, impaired response to growth inhibitory/differentiation signals, alterations in apoptosis, delayed or blocked senescence, prolonged angiogenesis, and the capacity for invasion and metastasis.84 Although one or more of these abnormalities can be detected at different stages of tumor progression and may thus be seen in premalignant lesions, all are present in advanced cancers. The driving force behind many of these changes is genomic instability, which facilitates the accumulation of mutations in both oncogenes and tumor suppressor genes that contribute to the observed aberrations in cell function.85 While some of these changes are cell-autonomous and can be studied in purified populations of tumor cells, others depend on various additional cell types recruited to participate in the development and progression of cancer in intact organisms. Both the intrinsic alterations in neoplastic keratinocytes and the influence of collaborating cell types in skin tumor biology are being elucidated through the use of powerful experimental models.
From analyses of both human SCC pathogenesis and experimental skin tumor induction by chemical carcinogens, specific premalignant and malignant stages have been identified that have typical phenotypic, genetic, and biochemical characteristics (Fig. 111-1).86,87 Note that in contrast to SCCs and most other cancers, BCCs do not appear to have precursor lesions or give rise to more aggressive cancers. This is likely related to the observation that the genomic instability that fuels neoplastic progression in other tumor types is largely missing in BCCs, even though a substantial proportion of these tumors harbor p53 mutations that would be expected to facilitate the accumulation of additional genetic defects. SCC starts as a single or small number of specific mutations in a cell that has the capacity to cycle, a change understood as the “initiation” of carcinogenesis. Upon clonal expansion, initiated cells form a premalignant lesion, such as a squamous papilloma in the mouse or an actinic keratosis in the human. Agents that enhance clonal expansion of initiated cells are called tumor promoters. Tumor promotion may occur as a consequence of exogenous exposures such as UVR, topical chemicals or medications, infections, or wound healing. Promotion may be an endogenous process influenced by diet, smoking, or immune suppression. The acquisition of additional mutations that provide a growth advantage to the incipient cancer cell may also serve as an autonomous promoting stimulus. Premalignant lesions undergo further phenotypic changes, often in a predictable sequence and commonly multifocal within a single lesion.88 Some foci or lesions progress at a faster rate than others, and these are at highest risk for malignant conversion.89 Premalignant progression encompasses the majority of the tumor latency period prior to malignant conversion, when the lesion shows invasive properties.
Initiation is usually a low frequency genetic event and is directly dependent on carcinogen dose. A large variety of carcinogen classes can initiate skin tumors in rodents (Table 111-3). The phenotype of initiated epidermal cells as defined from in vitro analysis includes a defect in maturation, escape from senescence, and an enhanced growth potential, but initiated cells are still responsive to negative regulation, for example by TGFβ. At the molecular level, initiation involves an alteration in signal transduction pathways that regulate cellular responses to extracellular signals, and these are internally regulated by proto-oncogenes and tumor suppressor genes.84,90 Tumor promotion is generally a nongenotoxic stimulus that results in a disturbance of tissue homeostasis.88 Multiple classes of agents have promoting properties in experimental skin carcinogenesis (Table 111-3
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


