CHAPTER 10 Lymphatic Malformations
KEY POINTS
Vascular malformations are mistakes in the development of the vascular system; hemangiomas are vascular tumors.
Endothelial hyperplasia is not a characteristic of vascular malformations.
Vascular malformations have multiple causes and complex clinical presentations; most vascular malformations are mixed.
Lymphoscintigraphy represents the highest standard in diagnostic tests for primary and secondary lymphedema (see Chapter 26).
Arteriography and venography are needed for patients who require surgical or endovascular treatment.
Surgical treatment is reserved for more complex forms of vascular malformations, especially those with low flow.
Lymphatic malformations are a type of morphostructural vascular disease. They have been described and classified in several ways because of their different clinical manifestations and types of onset.
Simple malformations are usually named in Latin (for example, hemangioma simplex, angioma telangiectaticum, and hemangioma cavernosum), whereas complex or mixed malformations are named for the authors who first described them (for example, Klippel-Trenaunay syndrome [leg length discrepancy, varicose veins, or erythema without evidence of arteriovenous fistulas] and Parkes-Weber syndrome [Klippel-Trenaunay syndrome with the presence of arteriovenous fistulas]). The incidence of vascular malformations is 1.5% worldwide 1 ; the prevalence of venous forms is 1 child in 5000 or 10,000, depending on the study. 2
In his 1876 treatise on the pathology of tumors, Virchow 3 discussed the first accurate medical description of a vascular malformation involving a cirsoid aneurysm, dating back to the sixteenth century. Nicoladoni 4 and Branham 5 nearly simultaneously described the occurrence of bradycardia after compression of a high-flow arteriovenous fistula. In 1964 Malan and Puglionisi 6 published the first useful classification of angiodysplasia. Szilagyi et al 7 further described clinical diagnosis and a therapeutic approach in 1976.
In articles published in 1982, Mulliken and colleagues 8 , 9 proposed a classification that distinguished between malformations or vascular abnormalities and vascular tumors (hemangiomas). This is currently the most widely used system. Vascular malformations are mistakes in the development of the vascular system; hemangiomas are vascular tumors. Mulliken et al 9 described the characteristics of the endothelium and the biology of vascular malformations and hemangiomas. Approximately 30% of hemangiomas are present at birth, and 70% occur in the first 3 months of life. They grow during the proliferative phase (usually the first year of life) and regress in subsequent years. The female/male ratio of hemangiomas is 5:1. In a biologic study, hemangiomas had endothelial hyperplasia and an increase in mast cell inclusions; the cells grew if cultured, and they incorporated 3H-thymidine in their proliferative phase. 10 Most hemangiomas regress spontaneously (95% are reduced by about 7 years of age) and require no therapy because any intervention (removal or reduction) would be aesthetically damaging due to healing of the tissues.
In contrast, endothelial hyperplasia is not a characteristic of vascular malformations. These lesions do not have a proliferative phase (that is, they do not grow if placed in culture) and do not incorporate 3H-thymidine. They showed no mast cell inclusions in the biologic study. 10 Their endothelium is surrounded by hyperplasia of the vascular wall. Approximately 90% of cases are present at birth, and the female/male ratio is 1:1. The growth of vascular malformations is related to the child’s growth and hemodynamic factors.
Embryology
VASCULAR SYSTEM
The first channels of the vascular system can be seen beginning in the third week of gestation (see Chapter 4, Embryology). In 1922 Woollard 11 sketched three stages of growth and differentiation:
Stage 1, the undifferentiated stage, is characterized by a small network of capillaries.
Stage 2, the retiform stage, involves the development of plexiform structures, increasing the vascular system’s volume and extent.
Stage 3, the maturation stage, occurs by the third week of gestation. The first large arteries, veins, and lymphatic ducts are seen. Vascular development is guided by receptors for growth factors.
The blood vessels begin to form on the seventeenth day in the extraembryonic regions, with the generation of blood islands in the mesoderm of yolk sac, the embryonic stalk, and chorionic villus. Subsequently, throughout the embryonic disc the vascular network gradually expands for vasculogenesis (the formation of the circulatory system) and angiogenesis (the sprouting of blood vessels from preexisting angioblastic cords), a gradual process. By the twenty-fourth day, the yolk sac is connected to the embryo through the two veins and three vitelline arteries (celiac trunk, superior mesenteric artery, and inferior mesenteric artery), and the red blood cells begin to circulate. The two allantoic veins and the two allantoic arteries pass through the pedicle, which will become the umbilical cord.
Various veins arise from the edge of the body of the embryo. These vessels are called common cardinal veins:
Two anterior cardinal veins from the cephalic endpoint
Two posterior cardinal veins from the tail endpoint
Two common cardinal veins, left and right
Initially the venous system is symmetrical, but during the second month of gestation the right component takes over. The inferior vena cava derives from the right vitelline vein, the left and right posterior cardinal veins, and the superior and inferior cardinal veins. The common cardinal veins, the allantoic veins, and the vitelline veins merge into the venous sinus that, incorporated into the right atrium, will include the orifices of the superior and inferior vena cava. The superior vena cava develops from the common cardinal vein (right branch).
LYMPHATIC SYSTEM
The lymphatic system derives in part from the mesoderm and in part from the mesenchyme, an undifferentiated tissue that retains the ability to transform into another type of tissue, even in adults.
The mesenchymal origin is evident in various cells. The reticular cells constitute the framework of the immunocompetent organs, particularly the lymph nodes, and can transform into macrophages. Endothelial cells, which line the inside of the capillaries and lymph vessels, are able to transform into macrophages.
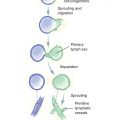
The origin of the lymphatic vessels is not entirely understood. They may develop directly from mesodermal protrusions or from endothelium evaginations of the veins in remodeling (lymphatic sacs). These structures are present in embryos measuring 2 cm (the fourth or fifth week of life or approximately 2 weeks after cardiovascular system development). Six primitive lymph sacs form:
Two equal and symmetrical sacs (jugular sacs) positioned cranially at the union of the jugular veins and the anterior cardinal veins
Two tail sacs (iliac sacs) at the union of the iliac veins and posterior cardinal veins
One median sac (retroperitoneal sac) on the posterior abdominal wall, at the root of the mesentery
One sac (cisterna chyli) dorsal to the retroperitoneal sac
The sacs are connected by lymphatic channels. Both main channels develop independently and are formed by primitive right and left thoracic ducts. They join caudally with an anastomotic branch, combining the jugular sacs with the cisterna chyli. In adults, the thoracic duct grows caudally from the distal portions of the primitive thoracic ducts (right and left) and cranially from left thoracic duct. The right lymphatic duct (large lymphatic right vein) derives from the cranial part of the right thoracic duct. The subclavian, jugular, and bronchomediastinal collecting trunks derive from the jugular sacs.
Classification of Vascular Malformations
Vascular malformations have multiple causes and complex clinical presentations, making their classification challenging. Malan and Puglionisi 6 tried to distinguish them by their anatomy and the presence or absence of arteriovenous fistulas. Szilagyi et al 7 divided them into three stages according to the three main phases of embryologic life (segmentation, gastrulation, and organogenesis). According to this classification, capillary malformations (hemangiomatous malformations) occur when development is blocked during the first embryologic phase. (These should not be confused with hemangiomas, which are skin tumors.) Microarteriovenous or macroarteriovenous fistulas occur when development is interrupted during the second embryologic phase. Persistent embryonic veins indicate a developmental block in the third embryologic phase and involve mixed segmental vascular malformations.
Jackson et al 12 based their classification on hemodynamic and angiography findings. High- or low-flow lesions are distinguished by the amount of blood flow in arteriovenous fistulas. High-flow malformations correspond to the presence of arteriovenous macrofistulas, and low-flow malformations correspond to arteriovenous microfistulas, which carry a better prognosis.
The most recent classification was published in Hamburg in 1990. 13 Rutherford et al 14 and Lee et al 15 later revised this classification (Table 10-1). Malformations are classified based on the predominant basic defect as follows:
Arterial
Venous
Lymphatic
Mixed, either truncal or extratruncal forms according to the flow along the main axis or branches
Predominantly arterial truncal malformations include congenital forms of sciatic artery or accessory subclavian artery persistence (which, passing behind the esophagus, is responsible for dysphagia lusoria); aortic arch anomalies; coarctation of the thoracic or abdominal aorta; and persistent embryonic vessels.
Predominantly venous truncal malformations include forms of persistent embryonic veins, such as the outer marginal vein. These present with mixed forms of aplasia, ectasia, aneurysm, and thrombosis of various anatomic segments. The most frequent alteration is ectasia of the venous system, which occurs in patients with Klippel-Trenaunay syndrome and makes up approximately 40% of the malformations. Venous aneurysms or hypoplasia of the venous wall is much less common, accounting for less than 10% of malformations.
Predominantly arteriovenous forms are most frequently extratruncal and may be localized or diffuse, especially on the skin or mucosa. They can be isolated or associated with more complex pathologic syndromes. Low-flow and high-flow shunt malformations can be distinguished. The predominantly arteriovenous malformations are more frequently seen in the lower limbs and pelvis, and the inherited forms usually occur in the lungs and sometimes in the brain. Hereditary hemorrhagic telangiectasia, for example, is characterized by the presence of spider veins in the dermis, mucosa, and viscera and is associated with arteriovenous fistulas in the lungs and brain. 16
These are manifested by recurrent nosebleeds, which occur in 90% of patients when they blow their nose or during a race or emotional stress. The mortality rate is 4%.
CAPILLARY MALFORMATIONS
Capillary malformations are also known as hemangiomas. They are most often found on the head and limbs (upper and lower). The dimensions are extremely variable, from localized forms to giant forms with involvement of an entire side of the body.
On clinical examination, capillary malformations appear as a red or rosy-purple skin rash with jagged or sharp margins. They are generally not raised, are highly variable in size and scope, and can be isolated or multiple and confluent. Within the patch, telangiectatic striae formed by larger-caliber capillaries are evident. In capillary-venous mixed forms, abnormal reticular veins that drain from the malformation can be seen. 17 , 18
Usually the skin lesion is sharply localized to the right or left of the midline, sometimes extending slightly beyond it. Another characteristic clinical aspect is metamerization (that is, the topography of the lesions usually follows the distribution of the head, trunk, or limb dermatomes). Facial locations typically respect the distribution of the sensory branches of the trigeminal nerve. In some patients, especially those with craniofacial lesions, hemangiomas tend to grow and produce a marked hyperplasia of the dermis and subcutaneous tissue. They are associated with a characteristic dilation of the subepidermal venous bed. These lesions are called hyperplastic birthmarks, commonly referred to as nevus flammeus or port-wine stains. On clinical examination, they present as patches of very dark purplish-red fibrotic tissue, often covered by vegetating plaques or polypoid formations. 19
LYMPHATIC MALFORMATIONS
Congenital malformations of the lymphatic system are characterized by embryogenetic capillary abnormalities or by malformations of the major lymphatic collectors of the limbs, head, and torso. 20 They are more common peripherally, especially in the legs, but can occur in the cervico-facial, thoracic, and pelvic regions. 21
According to the Hamburg classification, congenital malformations of the lymphatic system can be distinguished by tissue forms (extratruncal) and cysts forms (truncal forms or lymphangiomas). 22 Tissue lymphangiomas (or cystic hygromas) consist of a dense network of lymphatic microscopic vessels. Their dimensions are extremely variable, ranging from a small nodule to a voluminous mass. Lymphangiomas are characterized by an abnormal ectasia with saccular dilation of large lymph collectors or tanks. The locations correspond to the onset of major lymph node stations: submandibular, lateral cervical, axillary, inguinal, and mediastinal.
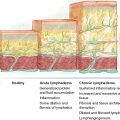
The clinical development of lymphatic malformations is closely related to the type and severity of anatomic abnormalities and the body region affected. The tissue lymphangiomas manifest as skin rashes or subcutaneous swellings with a whitish, warty, and irregular surface, often covered by translucent microvesicles containing serous fluid. Cystic hygromas occur as massive swellings under the skin and have a soft and spongy texture. They float and expand with moderate antigravity maneuvers and are nonpulsating.
The behavior of lymphatic malformations is extremely variable. They are usually present at birth and tend to gradually increase over the years, with remissions and relapses influenced by various factors (hormones, trauma, and infection). In some cases such as hemangiomas, progressive involution occurs after puberty. 23 , 24
Lymphatic malformations may manifest clinically with lymphedema, an edema with a particularly high-protein concentration that can lead to permanent disability. Lymphedemas are truncal lymphatic malformations, because they appear after an interruption in the anatomy of a lymphatic region. Truncal lymphatic malformations can develop at birth or later.
Lymphedemas are classified as primary or secondary. Primary lymphedema is connatal, early and late, and is often familial or inherited. Secondary lymphedema occurs after surgery, radiotherapy, trauma, and/or inflammation, or is functional (for example, postphlebitic). Secondary lymphedema is increasing in the average population because of the growing incidence of tumors. Treatments for tumors have improved; however, survival is associated with treatment sequelae, mainly secondary lymphedema.
Lymphodynamic impairment results from agenesis or aplasia of lymph nodes or lymphatic trunks, impaired permeability of lymph capillaries, or lymphadenodysplasia. It can be familial or inherited (for “sporadic mutations” in only one member of the family) or caused by chromosomal abnormalities, amniotic bands, or other congenital diseases. Prevention is essential. Therefore if familial, it is important to know the genetic mutation in order to study the same mutation in subjects with a blood relation with the proband who has the mutation.
The most frequent complications are usually local. Tissue forms can cause skin or mucosa necrosis, possibly associated with a lymphorrhage. Cystic forms can result in bleeding, infection, and compression of vital organs.
More than 70% of vascular malformations are mixed. 25 The most common forms seen clinically are listed in Table 10-2.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


