Breast Implants: Materials and Manufacturing Past, Present, and Future
William P. Adams Jr.
Jason K. Potter
Introduction
The female breast has long been an object of both individual and societal interest. The breast represents a woman’s femininity and sensuality. One does not have to look far in the media to recognize the relative value of the breast in female perception by society. These factors represent some of the underlying motives that have made surgery of the female breast, both aesthetic and reconstructive, an area of great interest and formidable challenge to plastic surgeons for decades. Arguably, no device or single surgical procedure has altered the landscape of breast surgery as dramatically as prosthetic breast implants. Breast implants have been in use since before the mid-1960s, yet the topic still produces heated debate and emotions. In November 2006, the U.S. Food and Drug Administration (FDA) lifted the 1992 ban placed on silicone implants, approving their use for breast reconstruction in women of all ages and breast augmentation in women older than 22 years of age. Despite their approval, many consumer advocacy groups oppose the decision, and the FDA has charged the manufacturers with the task of collecting 10-year data on 40,000 women to further evaluate their safety. This chapter outlines and discusses the past development of breast implants, as well as their present and future status.
Silicone
The building block of the breast implant is based on silicone science, and the biochemical principles are essential for developing implant technology. Silicone refers to a group of polymers based on the element silicon. Sand (silicon dioxide [SiO2]) is one of the most abundant compounds on earth. The polymer used medically is polydimethylsiloxane (PDMS) (Fig. 29.1). Silicones were first widely used in the World War II era for industrial and military applications. Medical applications for silicone were quickly identified, making silicone products commonplace within the medical industry today. Common silicone-containing products include indwelling catheters, extended wear contact lenses, pacemakers, syringes, and pharmaceuticals. Silicone products include industrial- and medical-grade materials. Medical grade refers to material that is pure and consistent in composition. Silicone is classified as a medical device, meaning that silicone does not achieve its primary intended purpose by chemical action or through its metabolism. Characteristics of silicone that are favorable for medical uses include thermal and oxidative stability, chemical and biologic inertness, hydrophobic nature, and sterilization capability.
Silicone polymers may be produced in a variety of forms, including oil, gels, or elastomers (rubber). The physical state is determined by the degree of chemical cross-linking. Cross-linking occurs between vinyl and hydrogen groups on silicon atoms. Silicone oils are straight chains of PDMS without cross-linking and are insoluble in water. PDMS will remain in liquid form indefinitely. Silicone gels consist of cross-linked (of various degrees) PDMS chains, together with variable amounts of PDMS liquid. It is important to keep in mind that the majority of silicone gel is silicone oil within the confines set by the PDMS gel matrix. The ratio of silicone liquid to gel is controlled by manufactures to control the viscosity of the gel. Elastomers of silicone have high degrees of cross-linking and almost no PDMS oil. Breast implant shells, both silicone and saline filled, consist of a vulcanized silicone elastomer that is reinforced with silica for increased strength (1). In an effort to reduce gel bleed from silicone-filled devices, phenyl or trifluoropropyl groups are bonded to the shell to decrease the shell permeability to PDMS oil (2,3). These “low-bleed” implant shells with “barrier coating” are characteristic of current third-, fourth, and fifth-generation implants (discussed later).
Manufacturing and Materials
Prior to the development of prosthetic breast implants numerous materials were put on trial for the purpose of augmenting the female breast. Until the 1950s, materials included autogenous fat and dermal grafts, fat injections, paraffin injections, and insertion of glass balls, ivory, rubber, and Terylene wool (4,5). These materials frequently led to infection, tissue necrosis, and firmness of the breast. The autogenous materials were uniformly troubled by resorption. The 1950s and 1960s saw the use of many other products. Free injections of hydrocarbons, petroleum jelly, silicone oils, vegetable oils, and bee’s wax were tried and led to many problems, including infection, granulomas, fistulas, and breast firmness (4,5). During this same time, many different types of sponges were developed in the hope that soft-tissue ingrowth would improve the biocompatibility of these products. Polyvinyl and polyether were the most common sponge materials (Fig. 29.2), whereas others included silastic and Teflon. Unfortunately, these products were compromised by many of the same problems and were discontinued.
The development of the silicone gel prosthesis in 1962 marked an important new era in breast surgery. Since Cronin and Gerow (6) first reported its use, few other materials have been used for breast augmentation. This is partly due to the success of silicone devices and also by the subsequent FDA regulation of medical devices that was started shortly after the development of silicone gel devices.
Having fewer materials has not been equated with having fewer devices. More than 200 different types of silicone breast implants and expanders have been manufactured in the United States (7). Since the original silastic gel implant was
produced, many modifications have been made to the design of these devices. The evolution in design and manufacturing of breast implants has focused on four major areas: characteristics of the shell, characteristics of the filler, shape, and surface configuration.
produced, many modifications have been made to the design of these devices. The evolution in design and manufacturing of breast implants has focused on four major areas: characteristics of the shell, characteristics of the filler, shape, and surface configuration.
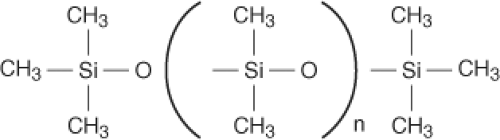 Figure 29.1. Polydimethylsiloxane. |
Any device can be expected to perform different from its counterparts based on its physical and structural properties, and this presents a significant challenge when we attempt to retrospectively review the outcomes of breast implant surgery since the mid-1960s. To assist in this review, an attempt has been made to classify breast implants into generations based on the time period of their development (8,9,10,11). This classification system recognizes the evolution and variations in the design that have occurred over time; however, it does not provide the ability to compare outcomes of breast implant surgery based on the time period the surgery was performed. Large overlaps of the periods of manufacturing of a particular device and the continued clinical use of those devices into the period of the next generation of manufacturing prevents this concept from having clinical applicability.
Implant Generations
When the generation scheme (Table 29.1) was first proposed, there were essentially three generations of breast implants corresponding to products developed in the 1960s (first generation), 1970s (second generation), and 1980s (third generation). First-generation devices are represented by the original silicone gel implant developed by Cronin and Gerow. This device, the Silastic 0, was manufactured by Dow Corning from approximately 1964 to 1968 (5). The Silastic 0 possessed a thick elastomer shell with seams and a viscous silicone gel. Dow Corning made several modifications to the original device, including changes in the elastomer, creating a seamless shell, and later made the shell much thinner. First-generation devices overall were characterized by thick shells, a thick viscous gel, and Dacron patches, and were produced until the late 1970s. The most commonly reported complication of these devices was capsular contracture.
 Figure 29.2. Polyether sponge used for breast augmentation. |
Table 29.1 Generations of Silicone Gel–Filled Breast Implants | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Second-generation devices were modified in an attempt to improve the rate of capsular contracture. These devices were designed with a much thinner shell (0.13 mm versus 0.25 mm average thickness) and a less viscous gel, and the Dacron patches were removed (5). The first second-generation device was Dow Corning’s Silastic I. It was introduced in 1972, and manufacturing of the Silastic I overlapped with the production of Silastic 0. Silastic I was produced until 1986. It did not provide any appreciable reduction in the incidence of capsular contracture and reportedly had a higher incidence of rupture that was attributed to the strength of its shell (5).
The phenomenon of gel bleed was realized in the 1970s (5,12,13,14). Gel bleed is the diffusion of non–cross-linked silicone oil from the gel across the elastomer shell into the surrounding environment. Although the significance of this phenomenon remains unclear, it stimulated manufacturing changes that are characteristic of third-generation devices. Thicker, reinforced barrier shells characterize third-generation devices. The thickness and strength improvements were developed out of concern for shell failure with second-generation devices. Shell strength was improved by reinforcing the elastomer composition with silica (1). Creating a barrier to gel diffusion with phenyl or trifluoropropyl groups bonded to the shell surface reduced diffusion of non–cross-linked silicone (2,3). These properties are retained in current manufacturing processes. It is important to keep in mind that gel bleed is a function of diffusion of silicone oil across the elastomer. The gel bleed does not change based on the viscosity (degree of cohesion of the gel filler).
Saline-filled breast implants were first manufactured in France in 1964, introduced by Arian with the goal of being surgically placed via smaller incisions. These devices had a high
failure rate and were discontinued in the early 1970s (5). Heyer-Schulte was the first U.S. manufacturer of saline-filled devices. The original devices consisted of thin shells created through a high-temperature vulcanization (HTV). These devices were prone to spontaneous deflation (5). Modifications in the shell manufacturing have allowed the high success rates that characterize modern saline-filled devices. The current devices are manufactured with thicker, room temperature–vulcanized (RTV) shells.
failure rate and were discontinued in the early 1970s (5). Heyer-Schulte was the first U.S. manufacturer of saline-filled devices. The original devices consisted of thin shells created through a high-temperature vulcanization (HTV). These devices were prone to spontaneous deflation (5). Modifications in the shell manufacturing have allowed the high success rates that characterize modern saline-filled devices. The current devices are manufactured with thicker, room temperature–vulcanized (RTV) shells.
Implant Filler
Modifications in the characteristics of the implant filler have also occurred. The most obvious is the change to saline-filled devices during the “implant crisis.” However, significant modifications have occurred in the silicone gel characteristics. The modifications in silicone gel technology are significant enough that many consider the modern era gels a fourth implant generation. Since 1992, due to increased demands to improve manufacturing processes, silicone gel implants have been improved devices with slightly thicker shells and more cohesive gel filler than third-generation devices.
Because breast implants are filled with medical-grade silicone, changes in silicone gel chemistry have centered on the cohesive quality of the gel. All silicone gels are cohesive, but the degree of cohesiveness has clinical importance. The degree of cohesiveness is a reflection of the elastic memory or shape retention of the gel. Cohesiveness is produced by the chemical cross-linking of the silicone gel molecules. The degree of cohesiveness imparts important characteristics to the structure and feel of the implant. Second-generation implants produced before 1985 contained minimally cohesive gels. Third- and fourth-generation devices evolved to contain increasingly cohesive gels after 1985, and in 1993, form-stable cohesive gel implants (Fig. 29.3) were introduced.
The fifth-generation implants are form-stable cohesive gel implants (e.g., Inamed 410 and Mentor CPG). These are shaped silicone gel devices with enhanced cohesion that offer improved breast shaping and results. These implants are currently undergoing clinical trials in the United States.
Silicone gel and saline are the only materials presently available for use as filling material for breast implants in the United States. Soy-filled implants (Trilucent) were marketed for a short time period in Europe but were voluntarily pulled from the market in 2000 by the manufacturer (15,16). Trilucent implants contained Trilipid 6, a medical-grade triglyceride fat extracted from soybean oil. This material was studied in animals and not shown to be a safety concern. Approximately 5,000 European women and 50 U.S. women received the implants as part of European and U.S. clinical trials. In the United States, the devices had limited availability through an investigation device exemption. The devices were taken out of clinical use due to the development of inflammatory reactions resulting from the leakage of the oil into the surrounding tissues (17,18). The reactions resolved with removal of the devices and did not present long-term health concerns. There are presently no other alternative fillers available through clinical trial.
 Figure 29.3. Cohesive gels consist of a highly cross-linked silicone that retains shape. |
Surface Texture
Surface texturing of silicone implants was first performed in the late 1960s, with the goal of preventing capsular contracture (5). Polyurethane-coated implants (Fig. 29.4) were introduced at this time but not popularized until the 1980s. Polyurethane devices were demonstrated to significantly lower the incidence of contracture, but two main concerns led to their discontinuation. Polyurethane was shown to undergo a degradation process in vivo that produced toluene diamine (19,20). This chemical was a known carcinogen in rodents and had unknown potential in humans (21). The risk for cancer has subsequently been shown to be extremely low in humans (22,23,24). Second, and probably more important, the polyurethane coating was shown to delaminate from the underlying silicone shell after several years in vivo (25). Preimplantation, the polyurethane implants were quite form stable; however, this was due to the properties of the stiff shell rather than the filler. The form retention was gradually lost as the polyurethane delaminated from the shell in vivo. Polyurethane-coated devices were voluntarily removed from the market in the early 1990s.
The effect of polyurethane on capsular contracture was theoretically attributed to its open cell structure, which allowed tissue ingrowth and prevented a regular, circumferential deposition of collagen, although a biochemical action at the cellular
level may have had an equal, if not a more important, effect. Manufacturers have attempted to develop new surfaces that perform as well as polyurethane (Fig. 29.5). Currently, two different textured silicone elastomer shells are available from Mentor Corporation and Inamed. In 1981, the American Society for Testing and Materials established norms for biocompatibility, mechanical properties of the shell, continence of the valves, modes of sterilization, packaging, and labeling. No guidelines for surface texturing are included, except the stipulation that surface texturing may not alter the other characteristics of the device. Mentor was the first to receive FDA approval for its textured shell. The Siltex pattern is created as a negative contact imprint of a texturing foam. This produces many fine nodules on the surface of the shell in a regular distribution. The size of these nodules ranges from 40 to 100 μm in height and from 70 to 150 μm in width (26). Inamed’s Biocell surface is produced through a lost salt technique. The implant shell is coated with finely graded salt under light pressure. The salt crystals are subsequently lost through the manufacturing process, leaving many fine depressions on the surface of the shell. These pores range from 600 to 800 μm in diameter and from 150 to 200 μm in height (26). Clinically, the Siltex texture is less aggressive than that of the Biocell; however, contrary to popular belief, true tissue ingrowth often does not occur with either texture unless the implant is placed in a tight pocket or in the tissue expansion environment (26). In regard to silicone gel implants, both surface textures have been shown to have benefits in improving the rate of capsular contracture, but this effect has not been realized in association with saline-filled devices, and therefore a universal benefit has not been demonstrated (5). Texturing also provides adhesiveness of the implant to the surrounding tissue. This is an extremely important consideration with shaped devices to prevent rotation and for certain considerations in breast reconstruction. Increased fold flaw failure (5,27) is a theoretical concern with a nonmobile device. When a device is not allowed to rotate, the natural folds in the elastomer shell do not cycle, and fatigue of the fold and subsequent failure may occur; however, this has not been demonstrated scientifically.
level may have had an equal, if not a more important, effect. Manufacturers have attempted to develop new surfaces that perform as well as polyurethane (Fig. 29.5). Currently, two different textured silicone elastomer shells are available from Mentor Corporation and Inamed. In 1981, the American Society for Testing and Materials established norms for biocompatibility, mechanical properties of the shell, continence of the valves, modes of sterilization, packaging, and labeling. No guidelines for surface texturing are included, except the stipulation that surface texturing may not alter the other characteristics of the device. Mentor was the first to receive FDA approval for its textured shell. The Siltex pattern is created as a negative contact imprint of a texturing foam. This produces many fine nodules on the surface of the shell in a regular distribution. The size of these nodules ranges from 40 to 100 μm in height and from 70 to 150 μm in width (26). Inamed’s Biocell surface is produced through a lost salt technique. The implant shell is coated with finely graded salt under light pressure. The salt crystals are subsequently lost through the manufacturing process, leaving many fine depressions on the surface of the shell. These pores range from 600 to 800 μm in diameter and from 150 to 200 μm in height (26). Clinically, the Siltex texture is less aggressive than that of the Biocell; however, contrary to popular belief, true tissue ingrowth often does not occur with either texture unless the implant is placed in a tight pocket or in the tissue expansion environment (26). In regard to silicone gel implants, both surface textures have been shown to have benefits in improving the rate of capsular contracture, but this effect has not been realized in association with saline-filled devices, and therefore a universal benefit has not been demonstrated (5). Texturing also provides adhesiveness of the implant to the surrounding tissue. This is an extremely important consideration with shaped devices to prevent rotation and for certain considerations in breast reconstruction. Increased fold flaw failure (5,27) is a theoretical concern with a nonmobile device. When a device is not allowed to rotate, the natural folds in the elastomer shell do not cycle, and fatigue of the fold and subsequent failure may occur; however, this has not been demonstrated scientifically.
 Figure 29.4. Polyurethane-coated silicone gel implant. |
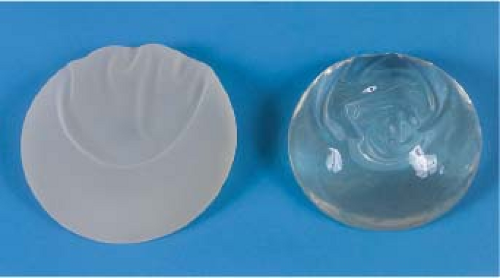 Figure 29.5. Textured and smooth surface gel implants. |
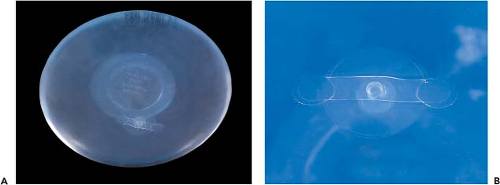 Figure 29.6. A, B: Diaphragm fill valve for saline-filled device. |
Valve technology for saline-filled devices has also evolved over time. The first Heyer-Schulte saline-filled implant used a Jenny valve. This was a diaphragm-type valve located on the anterior surface of the device. These valves were large and therefore easily palpable on the anterior surface of the device. Jenny valves were replaced in 1979 with the minidiaphragm valve (Fig. 29.6). These valves consist of a diaphragm valve on the anterior surface of the device. A fill tube with a small plastic tip is inserted into the valve, opening the diaphragm and allowing saline to enter the device. When the fill tube is removed, the device automatically closes. Other historical valves include the retention valve, which required a metal stylet to be placed through an anteriorly located valve; the leaf valve also used a metal stylet but accessed a posteriorly located valve. The spectrum valve was introduced by Mentor in 1992. It consists of a preplaced fill tube on the posterior side of the implant. Injection domes and connectors can be attached to the fill tube. The spectrum valve has a lower failure rate owing to three sealing mechanisms: kink, leaf, and plug. These three mechanisms effectively seal the inner and outer lumens of the valve.
In summary, there have been five generations of breast implants since their inception in the early 1960s. These generations are based on characteristics of the shell, characteristics of the filler, shape, and surface configuration. The clinical behavior of a given implant may be based on its respective generation.
Manufacturing Process
The manufacturing process for breast implants as with all medical devices is a tightly regulated process that must meet rigid criteria for quality assurance. Surprisingly, the majority of breast implant shells are produced individually through a handmade process (Figs. 29.7 and 29.8). Shells are produced through repeated dipping of mandrels into liquid silicone dispersion that is then cured with heat or humidity. After the silicone shell is cured, it may be removed from the mandrel, inspected for irregularities, and measured to ensure precise compliance with thickness standards. Shells are filled with their appropriate silicone gel and sealed with a patch bonded to the surface (Fig. 29.8). Saline-filled devices have a valve incorporated into the shell to allow filling of the device in vivo.
Breast Implants and the U.S. Food and Drug Administration
Introduced by Cronin and Gerow in 1962 (6), silicone gel breast implants redefined the modern era of breast augmentation. Use of silicone breast implants initially seemed the panacea for breast surgery, and the devices saw widespread use for several years. At the time silicone gel breast implants came on the market, the FDA did not regulate medical devices, and so the devices were marketed without having to go through clinical trials and without data to support their safety. Breast implants had been on the market for more than 10 years when, in 1976, the Medical Device Amendment to the Food, Drug,
and Cosmetic Act charged the FDA with regulating them as medical devices. The amendment provided a grandfather clause for devices considered equivalent to ones sold before 1976, and so both silicone and saline devices remained available (5,28). The amendment stipulated that manufacturers would be required to collect and submit safety and efficacy data to the FDA, and grandfathered devices would only remain on the market until the time when the FDA was able to formally evaluate their safety.
and Cosmetic Act charged the FDA with regulating them as medical devices. The amendment provided a grandfather clause for devices considered equivalent to ones sold before 1976, and so both silicone and saline devices remained available (5,28). The amendment stipulated that manufacturers would be required to collect and submit safety and efficacy data to the FDA, and grandfathered devices would only remain on the market until the time when the FDA was able to formally evaluate their safety.
Related posts:
 Follow-Up After Surgery for Primary Breast Cancer: Breast-Conserving Therapy and Mastectomy
Follow-Up After Surgery for Primary Breast Cancer: Breast-Conserving Therapy and Mastectomy
 One-Stage Immediate Breast Reconstruction with Adjustable Implants
One-Stage Immediate Breast Reconstruction with Adjustable Implants
 One-Stage Reconstruction of the Breast Using Autologous Tissue with Immediate Nipple Reconstruction
One-Stage Reconstruction of the Breast Using Autologous Tissue with Immediate Nipple Reconstruction
 Pedicled Perforator Flaps in Breast Reconstruction
Pedicled Perforator Flaps in Breast Reconstruction
 Management of Chronic Postoperative Breast Pain
Management of Chronic Postoperative Breast Pain
 The Periareolar Approach to Augmentation Mammaplasty
The Periareolar Approach to Augmentation Mammaplasty
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


