Fig. 13.1
Heme biosynthetic pathway (published in Dermatology, 3rd ed., Bolognia J and Gutierrez PAP, Porphyria, 718, Copyright Elsevier (2012))
Several PCT variants exist, including an acquired form denoted as “sporadic” or “Type 1 PCT” and an inherited autosomal dominant form denoted as “familial” or “Type 2 PCT.” A much less common familial variant, “Type 3 PCT,” is hypothesized to occur from the inheritance of an unidentified gene [2]. Irrespective of subtype, clinical manifestations typically occur when hepatic UROD activity decreases to 20 % of normal or less [3]. A variety of factors are known to precipitate PCT in all subtypes including environmental exposures, infections, nutritional influences, and other medical comorbidities.
The relationship between PCT and hemodialysis is well established in the scientific literature [4–28]. Though less common, there is also a reported association with peritoneal dialysis [29–32]. Furthermore, patients with impaired renal function, irrespective of dialysis treatment, may develop PCT due to inefficient excretion of porphyrin by-products [33].
Zelickson first described pseudoporphyria in 1964 after a patient taking nalidixic acid was observed to have the clinical findings of PCT, yet no porphyrin abnormalities were identified. Pseudoporphyria has indistinguishable clinical and histologic findings from PCT, but laboratory analysis does not demonstrate the abnormalities in porphyrin metabolism that defines PCT [34]. Pseudoporphyria has been classically reported in association with excessive exposure to ultraviolet A (UVA) from tanning beds, medication use, and in patients with chronic kidney disease.
In those patients with chronic renal disease, pseudoporphyria has also been termed “bullous dermatosis of end-stage renal disease” and is associated primarily with hemodialysis. Gilchrest first demonstrated the association between pseudoporphyria and hemodialysis in 1975, in a report of five patients presenting with PCT but exhibiting normal porphyrin levels [35]. Pseudoporphyria has also been observed in patients undergoing peritoneal dialysis as well as in patients with impaired renal function without exposure to dialysis [36–39]. Although it is a disorder characterized by normal porphyrin metabolism, dialysis patients with pseudoporphyria may exhibit mildly elevated serum porphyrin levels due to the poor porphyrin clearance associated with dialysis [19].
Epidemiology
While PCT is the most common of the porphyrias, it is considered to be an uncommon condition. The estimated prevalence of PCT is approximately 1 in 10,000 individuals [40]. Approximately 80 % of PCT patients have the sporadic (Type I) form, and the remaining 20 % are predominantly the familial (Type II) type. In both types, PCT most often presents during the third to fourth decade of life [41]. In the past, PCT demonstrated a greater frequency in men. However, current rates of PCT are approximately equal across both genders [41]. The increasing frequency among woman is hypothesized to reflect the greater use of oral contraceptives [41].
The incidence of PCT in patients undergoing hemodialysis has been reported to range from 1.2 to 18 % [18]. In the pre-erythropoietin era, the prevalence of PCT in dialysis patients may have been a more common occurrence due to the frequency of transfusion-related iron overload and perhaps transfusion-related hepatitis C infection [42].
A retrospective review of 20 patients with pseudoporphyria demonstrated a mean age of diagnosis to be 50 years, with a range of 33–71 years [43]. Though more common in adults, pseudoporphyria has been described in children. In a subset of children taking naproxen for joint pain associated with juvenile idiopathic arthritis, 11 % of naproxen users developed pseudoporphyria. The overall incidence and prevalence of pseudoporphyria is variable depending on the proposed inciting agent, such as medication use or ultraviolet A (UVA) exposure, but in all causes, fair skin appears to be an independent risk factor [45–48]. In patients with end-stage renal disease (ESRD) on hemodialysis, pseudoporphyria has been reported to occur at a rate from 1.2 % to as high as 18 %; pseudoporphyria occurs less frequently in those on peritoneal dialysis [49]. This discrepancy may be related to the lower levels of plasma uroporphyrin seen in patients receiving peritoneal dialysis [37].
Pathogenesis
Porphyria Cutanea Tarda
PCT is characterized as a multifactorial disorder related to either an inherited or acquired deficiency of UROD caused by both genetic and nongenetic factors. The abnormal physiology of PCT is related to the accumulation of porphyrins in the liver, plasma, and skin due to reduced activity of UROD. Approximately one-fifth of individuals with PCT have an inherited UROD mutation and exhibit UROD activity at 50 % of normal in all tissues from birth. The remaining majority of individuals with PCT have the sporadic form and exhibit reduced UROD activity only in hepatocytes. Porphyrin molecules have very intense absorption bands in the visible and ultraviolet A (UVA) electromagnetic spectrum especially in the Soret band, between 400 and 410 nm. Pathogenic porphyrin accumulation in the skin absorbs both visible and ultraviolet light, causing the main cutaneous signs and symptoms of PCT.
In both the sporadic and familial forms, the clinical expression of PCT requires additional susceptibility factors, genetic and/or environmental, to reduce UROD activity to 20 % of normal. In a study of 143 patients with PCT, the most frequent inciting agents included ethanol use, smoking, chronic hepatitis C virus (HCV) infection, and hemochromatosis HFE mutations; three or more susceptibility factors were recognized in 70 % of patients [50]. These proposed etiologic agents promote the accumulation of excess iron or oxidative stress in hepatocytes.
Hepatic iron overload is present in almost all cases of PCT, often with stores nearly twice that of normals. Iron has many proposed roles in the pathogenesis of PCT. Perhaps most importantly, iron directly inhibits UROD enzymatic activity and acts as a catalyst for the formation of reactive oxygen species that damages hepatic tissue [51]. Additional genetic associations with PCT that affect iron accumulation include hemochromatosis HFE gene mutations. Many patients with PCT, either genetic or acquired, are frequently found to have concurrent HFE gene mutations.
Alcohol has often been observed to precipitate PCT. Along with diminishing UROD activity after acute ingestion or in chronic alcoholics, ethanol may induce the enzyme aminolevulinic acid (ALA)-synthetase. This leads to porphyrin accumulation without the ability to complete the heme synthesis cascade due to inhibited UROD. Chronic alcoholism has also been associated with increased iron absorption and suppressed erythropoiesis.
Additional triggers reported to precipitate PCT include exposure to estrogens, environmental pollutants, human immunodeficiency virus (HIV) infection, and concurrent medical conditions including diabetes mellitus, systemic lupus erythematosus, hepatic steatosis, bone marrow disorders, and myeloproliferative disorders.
In patients with impaired renal function, PCT has several interconnected pathogenic mechanisms but in part results from impaired porphyrin clearance. Furthermore, patients with ESRD requiring renal replacement therapy may have even higher porphyrin levels since current dialytic techniques (hemodialysis or peritoneal dialysis) are substandard in clearing porphyrins from circulation [33]. Indeed, porphyrin clearance across the dialysis membrane in a hemodialysis patient with PCT was demonstrated to be 100 times less than the renal clearance of porphyrin molecules in PCT patients with normal renal function [20]. Furthermore, individuals undergoing chronic hemodialysis are at an increased risk of developing acquired forms of UROD deficiency due to hepatotoxicity from various exposures, such as alcohol, statins, estrogens, viruses, or iron overload [19]. Other mechanisms proposed to induce abnormal porphyrin metabolism in dialysis patients include the reduction of UROD from azotemia [52], exposure to phototoxic medications, and aluminum load [53].
Pseudoporphyria
Although the mechanism is not fully understood, pseudoporphyria has been associated independently with various medications, excessive sun exposure, ultraviolet A (UVA) radiation, chronic kidney disease, and dialysis [49]. Additional reported associations include phototherapy with ultraviolet B (UVB), hormone replacement, human immunodeficiency virus (HIV) and HCV infections, and other coexisting medical conditions including systemic lupus erythematosus, vitiligo, sarcoidosis, Sjögren syndrome, and hepatoma.
The pathogenesis of pseudoporphyria in individuals with chronic kidney disease is not fully established. However, several mechanisms have been proposed and may not be mutually exclusive. These theories have included a primary drug-induced pathogenesis, as this population is often concurrently managed with many drugs known to be associated with pseudoporphyria, such as nifedipine, furosemide, and erythropoietin; an increase in oxidative stress due to dialysis-associated glutathione depletion; and in those patients undergoing dialysis, a potential role of aluminum hydroxide that is found in the dialysate and that has demonstrated phototoxicity in animal models [49]. It is important to note that the association between erythropoietin and pseudoporphyria may be of minor significance as pseudoporphyria appeared in the literature prior to the introduction of erythropoietin [49]. Both peritoneal dialysis and hemodialysis reduce glutathione levels in the blood and erythrocytes; the resultant glutathione deficiency creates an increased susceptibility to oxidative stress in these patients and may facilitate photo-oxidative cutaneous injury [54, 55]. The development of pseudoporphyria in nondialyzed individuals with chronic kidney disease has been reported in the scientific literature, suggesting that renal dysfunction may play a more significant role in the induction of pseudoporphyria than previously hypothesized; however, there may have also been a drug-induced component lending evidence to the multifactorial mechanisms in this patient population [37].
Clinical
The predominant clinical characteristics of all forms of PCT usually involve photocutaneous and liver manifestations. There are several characteristic skin findings, but the most commonly observed features include skin fragility and blistering on the hands, present in the vast majority of cases (Fig. 13.2a, b) [56]. Along with the observable skin changes, a notable symptom reported frequently by patients is increased pruritus of the affected skin. Though many uremic patients have chronic pruritus, the development of PCT may heighten symptoms. Other classic features of PCT include facial hypertrichosis, most notably on the temples (Fig. 13.3a, b); photodistributed hyperpigmentation; premature aging; and milial cysts and comedones (Fig. 13.4). Less common, but still characteristic, findings include sclerodermoid changes, facial blisters, scarring alopecia, and photo-onycholysis (Fig. 13.5) [56, 57]. Because of the underlying porphyrin accumulation, patients may report the darkening of their urine. This finding may not be as specific in oliguric or anuric patients. In PCT, the liver is the other predominant organ system affected. The range of hepatic findings is diverse and may include steatosis, fibrosis, siderosis, and as a late manifestation, cirrhosis [58]. Complete hepatic evaluation must be performed as these patients may be at risk for an underlying hepatocellular carcinoma [59]. These liver findings often relate to the underlying inciting PCT trigger, such as HCV, alcohol abuse, iron overload, or hemochromatosis. There may be additional clinical findings more specific to these conditions as opposed to being directly related to PCT [60].
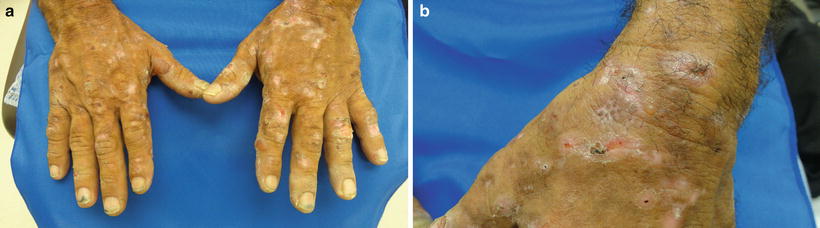
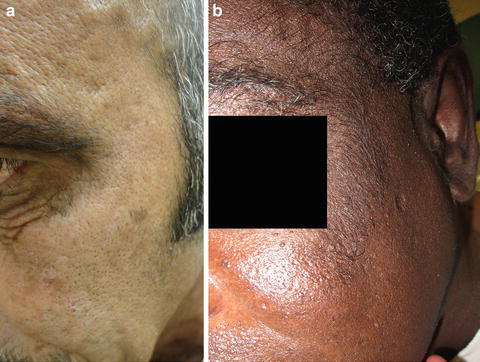
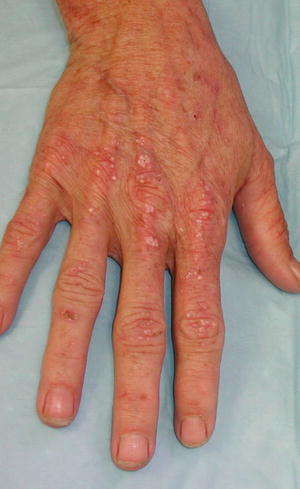
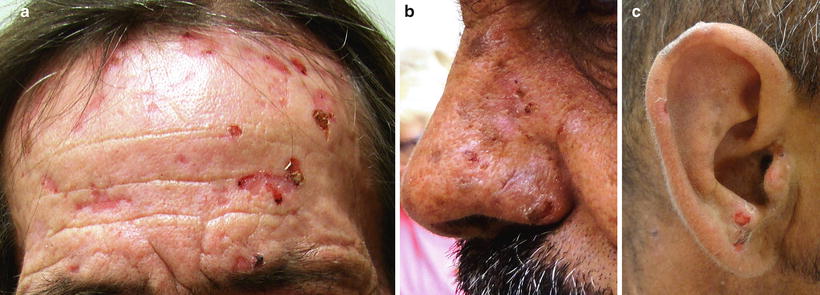
Fig. 13.2
PCT classically presents with skin fragility and vesicles on the dorsal surface of hands
Fig. 13.3
Hypertrichosis is another characteristic feature of PCT and can involve the forehead, temples, and upper cheeks
Fig. 13.4
Milia formation is a common occurrence following extensive blistering in PCT. Courtesy of Julia R. Nunley, M.D.
Fig. 13.5
Photo-onycholysis, a sun-induced nail dystrophy, is seen along with erosions and milial cysts on the bilateral dorsal hands
The clinical characteristics of pseudoporphyria greatly resemble PCT. In pseudoporphyria, the most frequent clinical characteristic is photodistributed blistering that occurs in nearly every patient. Also characteristically observed are scarring, photosensitivity, and skin fragility. Though not as common, milia may develop and are often directly related to the presence of scarring [43]. In contrast to PCT, noticeably absent clinical features include hypertrichosis, hyperpigmentation, and sclerodermoid changes [42].
Differential Diagnosis
The differential diagnosis for PCT includes other forms of the porphyrias (including variegate porphyria, congenital erythropoietic porphyria, hepatoerythropoietic porphyria, and hereditary coproporphyria), pseudoporphyria, other bullous skin disorders (including bullous pemphigoid, bullous drug eruption, epidermolysis bullosa, epidermolysis bullosa acquisita, and bullous lupus erythematosus), and hydroa vacciniforme, a rare chronic photodermatosis.
Laboratory analysis of urinary and fecal porphyrin levels allows for differentiation of PCT and pseudoporphyria from other subtypes of porphyrias [41]. To distinguish PCT and pseudoporphyria from other vesiculobullous disorders, a skin biopsy for histology and direct immunofluorescence is recommended [42]. Immunohistochemical analysis of tissue submitted for direct immunofluorescence would differentiate autoimmune immunobullous disorders from PCT. Genetic immunobullous diseases can generally be differentiated based on time of onset, as most are present at or near birth. Hydroa vacciniforme is considered a disease of childhood, is associated with Epstein–Barr virus, and rarely has hepatic involvement.
Workup
The workup for PCT and pseudoporphyria includes clinical data, laboratory analysis, imaging studies, and histopathologic data. PCT and pseudoporphyria frequently simulate various subepidermal bullous disorders; thus, complete integration of clinical, histopathologic, and biochemical information is necessary for accurate diagnosis. Of note, results from 24 h urinary collections in the setting of significantly low renal clearance, especially in those who are oliguric or anuric, are not reliable.
Histology
PCT has various clinical findings, but a punch biopsy of the edge of photodistributed vesicles often demonstrates the pathognomonic features that aid in diagnosis. Histology of these vesicobullae demonstrates a cell-poor subepidermal bullae with festooning of dermal papillae, minimal inflammatory infiltration, and periodic acid-Schiff (PAS)-positive thickening of the papillary vessel wall [61] (Fig. 13.6). Intraepidermal eosinophilic collections of basement membrane type IV collagen, denoted as “caterpillar bodies,” are frequently observed [62]. Direct immunofluorescence frequently demonstrates linear IgG, C3, and fibrinogen along the dermal perivascular area and dermoepidermal junction [42], with less frequent observations of IgA and IgM deposition [63].
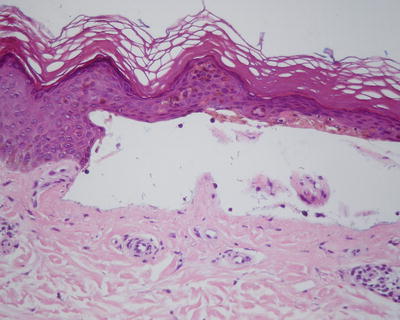
Fig. 13.6
Cell-poor subepidermal bullae with festooning of dermal papillae
The histologic profile of pseudoporphyria is nearly identical to PCT and cannot be reliably used to differentiate the two disorders. One minor difference reported is less blood vessel wall thickening with PAS staining in pseudoporphyria as compared to cases of PCT [61].
Laboratory Abnormalities
PCT is a disorder characterized by abnormalities in biochemical porphyrin levels. The urinary porphyrin profile demonstrates marked elevations in uroporphyrin I with less significant elevations in uroporphyrin III, 8-carboxyl uroporphyrin, and 7-carboxyl porphyrin [61]. Additionally, plasma uroporphyrin and fecal isocoproporphyrin IIII demonstrate elevations [42]. The urinary porphobilinogen (PBG) level is normal and the ALA level may be slightly elevated [3]. Erythrocyte porphyrin levels are normal or modestly elevated. It is important to note that uroporphyrin levels in asymptomatic ESRD patients may resemble or exceed levels found in individuals with PCT and normal renal function, as uroporphyrins are not sufficiently excreted via hemodialysis [65].
In anuric patients, fecal analysis for elevated levels of isocoproporphyrin III and plasma analysis for uroporphyrin are important diagnostic measures [32]. Fecal analysis remains the most accurate diagnostic marker of PCT in patients with chronic kidney disease as plasma uroporphyrin levels normally exhibit marked elevations due to impaired renal and dialytic clearance [64].
Additionally, workup for hemochromatosis may demonstrate positive results, given the strong association between hereditary hemochromatosis HFE gene mutations and PCT [66]. For the diagnosis of familial PCT, genetic sequencing of the UROD gene is necessary and is typically present as a heterozygous mutation.
Because liver involvement frequently accompanies PCT, investigation of hepatic activity is recommended. Laboratory tests include complete blood count, serum ferritin, serum iron, liver function profile, and screening for HCV, HBV (hepatitis B virus), and HIV infections. Most patients with PCT exhibit iron overload, which can be observed by high normal or moderate elevations in serum iron and ferritin levels. An analysis of serum alpha-fetoprotein is beneficial in screening for hepatocellular carcinoma [67].
Another diagnostic measure for PCT includes examination of urine under Wood lamp illumination in the dark, but after exposure to natural light (Fig. 13.7). If adequately concentrated, the urine of PCT patients will exhibit a pink to red fluorescence under a ultraviolet A (UVA) source and will turn red to brown after exposure to natural light for several hours [41]. However, it is important to note that these tests are neither sensitive nor specific.
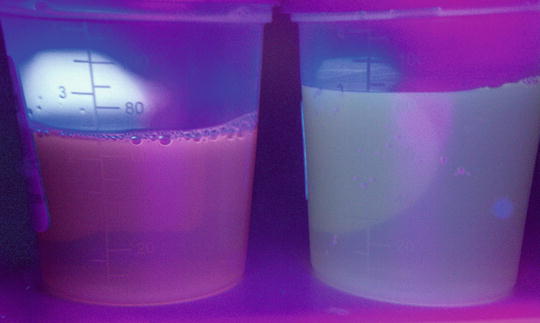
Fig. 13.7
Pink fluorescence of patient urine (left) compared to normal urine (right) under a Wood lamp illumination
Pseudoporphyria is a disorder characterized by normal biochemical porphyrin levels in the serum, urine, and stool. The diagnosis of pseudoporphyria in ESRD patients on dialysis is challenging because many of these individuals exhibit elevated plasma porphyrins despite having normal porphyrin metabolism [68]. Additionally, when comparing uroporphyrin levels in dialysis patients, hemodialysis patients frequently exhibit greater plasma uroporphyrin levels than patients undergoing peritoneal dialysis [37]. Thus, mildly elevated serum porphyrin levels should be anticipated in dialysis patients with bullous lesions who are diagnosed with pseudoporphyria [19]. Plasma porphyrin and fecal porphyrin levels should be assayed in ESRD patients, as urinary levels may be inaccurate. Liver enzyme analysis for individuals with pseudoporphyria is most often normal; however, abnormalities have been observed [49].
Imaging Studies
Imaging of the liver may be recommended in certain cases of PCT to evaluate hepatic iron content, size, and hepatic carcinoma.
Treatment
Porphyria Cutanea Tarda
Primary management of PCT in renal disease involves a combination of eliminating or reducing triggers, reducing iron load, and removing excess porphyrin accumulation. In some refractory cases, kidney transplantation may be a consideration.
Trigger Avoidance
If a known trigger of PCT has been identified, every effort should be made to minimize or eliminate the causative agent. Estrogen use should be discontinued, at least until complete remission is achieved, and attempts should be made to avoid hepatotoxic drugs. Active viral disease, such as with HIV and HCV, should be identified and appropriate therapy for these infections should be a part of the management plan. Sun protection is often of paramount importance. Beyond sun avoidance and protective clothing, ultraviolet A (UVA)-absorbing sunscreens, such as those containing avobenzone or zinc oxide, may be the best option when patients do plan to be exposed. Other lifestyle changes that should be encouraged include smoking cessation and strict alcohol avoidance.
Reducing Iron Load
Phlebotomy
After exogenous exacerbating factors are removed or discontinued, medical therapies employed to reduce iron load are the treatment of choice [69]. Even in patients with ESRD on either hemodialysis or peritoneal dialysis, the first line of treatment is therapeutic phlebotomy, as long at the hemoglobin is adequate. By using phlebotomy to reduce iron stores, the heme synthetic pathway normalizes both by augmenting UROD activity, an enzyme that is inhibited by iron, and through regeneration of new hepatic UROD [70]. Caution must be exercised with repeated phlebotomies in patients with renal disease due to the challenge of concurrent chronic anemia that is frequently observed. Most patients with renal failure are unable to tolerate the standard protocol of once or twice weekly removal of 500 mL of blood. Instead, small-volume phlebotomy, ranging from 100 to 350 mL, every 2–3 weeks is employed while aiming for target serum hemoglobin between 10 and 11 g/dL. Nevertheless, both standard volume and small-volume phlebotomy have induced clinical remission in PCT patients associated with chronic kidney disease [21, 22, 30].
For those patients with severe anemia, the use of recombinant human erythropoietin in conjunction with small-volume phlebotomies has been a successful strategy [14, 69–72]. By stimulating erythropoiesis, tissue iron is mobilized and circulating erythrocyte numbers are increased to levels that permit ongoing phlebotomy. When used concurrently with phlebotomy, high dose erythropoietin (150 U/kg three times per week) has been reported to be more efficacious than low dose (50 U/kg two to three times weekly) [70] Interestingly, successful anecdotes of erythropoietin monotherapy (150 U/kg three times weekly) have been reported in hemodialysis-related PCT [10–12, 73]. Phlebotomy is discontinued once serum iron and ferritin levels reach slightly low to low-normal values.
Deferoxamine
In patients with severe anemia or who are unable to undergo phlebotomy, use of deferoxamine, a ferrous iron chelator, may be an alternative. Deferoxamine has demonstrated measurable success in the scientific literature as a treatment method for dialysis-related PCT [6, 15, 23, 24, 31]. Remission of hemodialysis-related PCT has been reported following treatment with 40 mg/kg intravenous deferoxamine every week for 6 weeks [24]. There is one report of low dose hydroxychloroquine (400 mg weekly) used in conjunction with 40 mg/kg deferoxamine intravenously every week for 6 weeks in a patient with chronic dialysis-related PCT. Resolution of symptoms occurred within six cycles of treatment [6].
Reducing Porphyrins
Antimalarials
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


