Biology of Melanocytes: Introduction
|
Embryonic Development
Melanocytes are pigment-producing cells that originate from the dorsal portions of the closing neural tube in vertebrate embryos1 (eFig. 72-0.1). They derive from pluripotent neural crest cells that differentiate into numerous cell lineages including neurons, glia, smooth muscle, craniofacial bone, cartiledge, and melanocytes.2,3 Progenitor melanoblasts migrate dorsolaterally between the mesodermal and ectodermal layers to reach their final destinations in the hair follicles and the skin as well as inner ear cochlea, choroids, ciliary body, and iris.2,4 Pigment-producing cells can be found in fetal epidermis as early as the 50th day of gestation.
eFigure 72-0.1
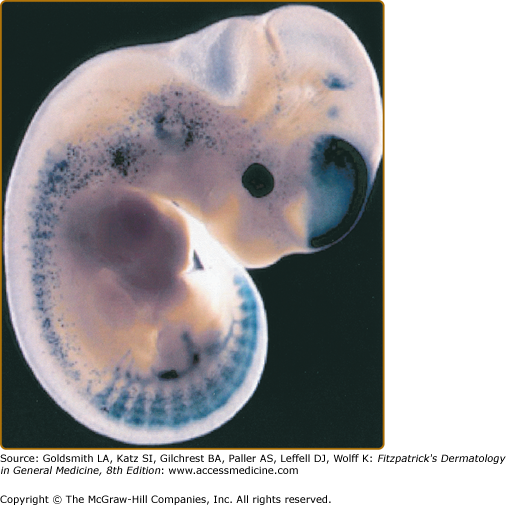
Migration of melanoblast precursors. Migrating melanoblasts in an E11.5 transgenic mouse embryo expressing the Lac Z reporter gene under the control of the melanocyte-specific Dct (DOPAchrome tautomerase) promoter. The melanoblasts are visualized by staining for β-galactosidase. (Used with permission from Ian Jackson and Alison Wilkie, Medical Research Council, Human Genetics Unit, Western General Hospital, Edinburgh, Scotland.)
Melanoblast migration and differentiation into melanocytes are influenced by a number of signaling molecules produced by neighboring cells. These include Wnt, endothelin (ET)-3, bone morphogenetic proteins (BMPs), steel factor (SF) (stem cell factor, c-Kit ligand), and hepatocyte growth factor (HGF/scatter factor).5–10 By interacting with their specific cell surface receptors, these molecules induce intracellular and intranuclear signaling to influence gene transcription and protein synthesis. Genetic defects in some of these molecules are associated with human genetic diseases: ETs (Waardenburg syndrome and Hirschsprung disease) and c-Kit and stell factor with piebaldism. Detailed discussion of each signaling molecule is available online.
 Wnt is expressed in the dorsal neural tube during neural crest cell migration and directs the maturation of pluripotent neural crest cells into melanoblasts.
Wnt is expressed in the dorsal neural tube during neural crest cell migration and directs the maturation of pluripotent neural crest cells into melanoblasts.
 The Wnt family is composed of 16 different secreted glycoproteins. They bind and activate Frizzled, a transmembrane heptahelical G protein-linked receptor,11 and induce the accumulation of β-catenin (eFig. 72-0.2A). Under baseline conditions, when Wnt does not bind Frizzled, cytosolic β-catenin is complexed with the enzyme glycogen synthase kinase 3β (GSK3β) that induces rapid ubiquitin-mediated degradation of β-catenin by cellular proteosomes. Binding of Wnt to its receptor Frizzled inhibits GSK3β activity, leading to β-catenin accumulation in the cytosol followed by its translocation to the nucleus (eFig. 72-0.2B). In the nucleus, β-catenin binds specific transcription factors and together the complex induces the transcription of microphthalmia-associated transcription factor (MITF).10 MITF affects melanoblast differentiation by inducing the transcription of three enzymes that regulate melanin synthesis: (1) tyrosinase, (2) tyrosinase-related protein-1 (TRP-1), and (3) DOPA (TRP-2).12
The Wnt family is composed of 16 different secreted glycoproteins. They bind and activate Frizzled, a transmembrane heptahelical G protein-linked receptor,11 and induce the accumulation of β-catenin (eFig. 72-0.2A). Under baseline conditions, when Wnt does not bind Frizzled, cytosolic β-catenin is complexed with the enzyme glycogen synthase kinase 3β (GSK3β) that induces rapid ubiquitin-mediated degradation of β-catenin by cellular proteosomes. Binding of Wnt to its receptor Frizzled inhibits GSK3β activity, leading to β-catenin accumulation in the cytosol followed by its translocation to the nucleus (eFig. 72-0.2B). In the nucleus, β-catenin binds specific transcription factors and together the complex induces the transcription of microphthalmia-associated transcription factor (MITF).10 MITF affects melanoblast differentiation by inducing the transcription of three enzymes that regulate melanin synthesis: (1) tyrosinase, (2) tyrosinase-related protein-1 (TRP-1), and (3) DOPA (TRP-2).12
eFigure 72-0.2
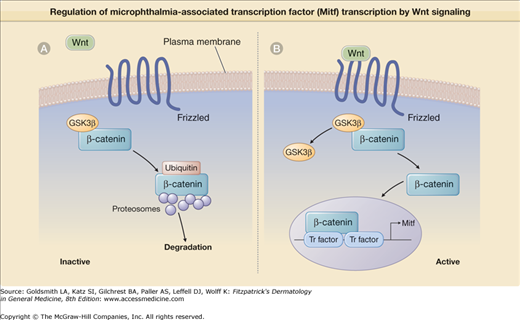
Regulation of microphthalmia-associated transcription factor (MITF) transcription by Wnt signaling. A. When Wnt is not bound to Frizzled, a membrane-spanning domain receptor, Frizzled is inactive. Under these inactive conditions, glycogen synthase kinase 3β (GSK3β) is complexed with β-catenin, enhancing its ubiquitin-mediated degradation by cellular proteosomes. B. Wnt binding activates Frizzled, leading to GSK3β dissociation from β-catenin. β-Catenin then translocates to the nucleus, where it binds with specific transcription factors (Tr factor) to initiate MITF transcription.
 Bone morphogenetic proteins (BMPs) are disulfide-linked dimeric proteins, produced as large precursors.13 They include more than 20 secreted proteins all sharing amino acid homology.14 BMPs belong to the transforming growth factor-β (TGF-β) family of secreted growth factors and their signaling suppresses neural crest cell differentiation into melanoblasts and thus may be viewed as Wnt antagonists. Accordingly, there is a decrease in BMP expression in the dorsal neural tube at the time of melanoblast migration.8
Bone morphogenetic proteins (BMPs) are disulfide-linked dimeric proteins, produced as large precursors.13 They include more than 20 secreted proteins all sharing amino acid homology.14 BMPs belong to the transforming growth factor-β (TGF-β) family of secreted growth factors and their signaling suppresses neural crest cell differentiation into melanoblasts and thus may be viewed as Wnt antagonists. Accordingly, there is a decrease in BMP expression in the dorsal neural tube at the time of melanoblast migration.8
 Endothelins (ETs) are a family of peptides, 21 amino acids long, originally identified by their vasoactive properties that include three members: (1) ET-1, (2) ET-2, and (3) ET-3.15 They are produced via proteolysis of larger precursor molecules. Like Wnt, ETs bind and activate heptahelical transmembrane G-protein-linked receptors EdnrA and EdnrB. ET-3, which is synthesized by ectodermal cells, and its EdnrB receptor, are particularly important during melanoblast migration along the dorsolateral pathway15 and their proper expression is required for survival, proliferation, and/or migration of melanoblasts.
Endothelins (ETs) are a family of peptides, 21 amino acids long, originally identified by their vasoactive properties that include three members: (1) ET-1, (2) ET-2, and (3) ET-3.15 They are produced via proteolysis of larger precursor molecules. Like Wnt, ETs bind and activate heptahelical transmembrane G-protein-linked receptors EdnrA and EdnrB. ET-3, which is synthesized by ectodermal cells, and its EdnrB receptor, are particularly important during melanoblast migration along the dorsolateral pathway15 and their proper expression is required for survival, proliferation, and/or migration of melanoblasts.
 Defects in either ET-3 or its EdnrB receptor result in prominent melanocyte loss.16 It appears that the receptor plays a more critical role in melanocyte development as compared to its ligand, as ET-3-null mice do not have as severe depigmented phenotype as EdnrB-null mice. Since ETs and Ednrs also affect the development of neural crest cells other than melanoblasts, defects in ET-3 or EdnrB lead to disease symptomology beyond pigmentary disorders, such as that present in type IV Waardenburg syndrome and in Hirschsprung syndrome16 (see Chapter 72).
Defects in either ET-3 or its EdnrB receptor result in prominent melanocyte loss.16 It appears that the receptor plays a more critical role in melanocyte development as compared to its ligand, as ET-3-null mice do not have as severe depigmented phenotype as EdnrB-null mice. Since ETs and Ednrs also affect the development of neural crest cells other than melanoblasts, defects in ET-3 or EdnrB lead to disease symptomology beyond pigmentary disorders, such as that present in type IV Waardenburg syndrome and in Hirschsprung syndrome16 (see Chapter 72).
 Early melanoblast development requires the presence of the cytokine steel factor (SF) (mast/stem cell factor, c-Kit ligand) and its tyrosine kinase transmembrane receptor c-Kit.17 SF is expressed by epidermal keratinocytes and as soon as c-Kit is expressed on melanoblasts, they begin their migration to their final destination.18
Early melanoblast development requires the presence of the cytokine steel factor (SF) (mast/stem cell factor, c-Kit ligand) and its tyrosine kinase transmembrane receptor c-Kit.17 SF is expressed by epidermal keratinocytes and as soon as c-Kit is expressed on melanoblasts, they begin their migration to their final destination.18
 Piebaldism is an autosomal dominant disorder that results from mutations of c-Kit or SF and leads to melanoblast failure to migrate to the skin and/or survive there19,20 (see Chapter 72). Affected individuals display broad depigmented patches, most prominent on the central forehead and trunk. Interestingly, the ventral aspect of the body is more frequently affected than the dorsal aspect, supposedly because it is the area farthest from the dorsally located neural crest where melanoblast migration begins.19,21 Complete absence of melanocytes as well as abnormalities of the reproductive and hematopoietic systems whose development also depends on SF/Kit receptor, have been observed in mice with homozygous loss of SF or c-Kit.22,23
Piebaldism is an autosomal dominant disorder that results from mutations of c-Kit or SF and leads to melanoblast failure to migrate to the skin and/or survive there19,20 (see Chapter 72). Affected individuals display broad depigmented patches, most prominent on the central forehead and trunk. Interestingly, the ventral aspect of the body is more frequently affected than the dorsal aspect, supposedly because it is the area farthest from the dorsally located neural crest where melanoblast migration begins.19,21 Complete absence of melanocytes as well as abnormalities of the reproductive and hematopoietic systems whose development also depends on SF/Kit receptor, have been observed in mice with homozygous loss of SF or c-Kit.22,23
 Hepatocyte growth factor (HGF/scatter factor) is the ligand for the transmembrane tyrosine kinase receptor Met. In vitro studies performed on murine neural crest cells show that HGF induces melanoblast proliferation and allows their differentiation into mature melanocytes.24 In addition, HGF regulates cadherin expression in melanocytes, specifically downregulating E-cadherin, thus affecting melanoblast homing (see below). Interestingly in mice, Met-null mutations do not appear to affect melanoblast number or migration and no decrements in Met function have been identified to date in human pigmentary disorders. Thus, the role of HGF/Met signaling during melanocyte development is still unclear.
Hepatocyte growth factor (HGF/scatter factor) is the ligand for the transmembrane tyrosine kinase receptor Met. In vitro studies performed on murine neural crest cells show that HGF induces melanoblast proliferation and allows their differentiation into mature melanocytes.24 In addition, HGF regulates cadherin expression in melanocytes, specifically downregulating E-cadherin, thus affecting melanoblast homing (see below). Interestingly in mice, Met-null mutations do not appear to affect melanoblast number or migration and no decrements in Met function have been identified to date in human pigmentary disorders. Thus, the role of HGF/Met signaling during melanocyte development is still unclear.
 Cadherins are a family of transmembrane glycoproteins (E-, P-, and N-) that promote calcium-dependent cell-to-cell adhesion.25 In melanoblasts, their cytoplasmic domains bind β-catenin. Murine studies show that E-cadherin is induced in melanoblasts prior to their entry into the epidermis. Later, when melanoblasts migrate into the hair follicle, E-cadherin expression is muted and melanoblasts begin to express P-cadherin.26,27 It is thought that coordinate expression of E-cadherins by epidermal keratinocytes and melanocytes plays a role in suppressing melanocyte proliferation in the epidermis. Interestingly, at times, some epidermal melanocytes switch from E-cadherin to N-cadherin and this switch allows them to escape the keratinocyte-mediated growth suppression, and proliferate/aggregate in nests to form nevocellular nevi.28 Generally, cadherin expression by melanocytes matches cadherin expression of surrounding cells.
Cadherins are a family of transmembrane glycoproteins (E-, P-, and N-) that promote calcium-dependent cell-to-cell adhesion.25 In melanoblasts, their cytoplasmic domains bind β-catenin. Murine studies show that E-cadherin is induced in melanoblasts prior to their entry into the epidermis. Later, when melanoblasts migrate into the hair follicle, E-cadherin expression is muted and melanoblasts begin to express P-cadherin.26,27 It is thought that coordinate expression of E-cadherins by epidermal keratinocytes and melanocytes plays a role in suppressing melanocyte proliferation in the epidermis. Interestingly, at times, some epidermal melanocytes switch from E-cadherin to N-cadherin and this switch allows them to escape the keratinocyte-mediated growth suppression, and proliferate/aggregate in nests to form nevocellular nevi.28 Generally, cadherin expression by melanocytes matches cadherin expression of surrounding cells.
Site-Specific Melanocytes
Generally, stem cells are defined by their undifferentiated state and their capacity to develop into several differentiated cell types. They are quiescent, slow-cycling cells that frequently are found in niches where they are surrounded by differentiated cells that affect their behavior through the secretion of cytokines and growth factors.29,30
Melanocyte stem cells reside in the hair follicle bulge (Fig. 72-1). They express TRP-2 as well as the neural crest stem cell intermediate filament nestin in addition to other neural crest stem cell markers including the transcription factors Sox10 and Pax5 that participate in the regulation of microphthalmia-associated transcription factor (MITF) and TRP-2. Melanocyte stem cells can leave the bulge region and migrate/differentiate in the epidermis or the hair follicle.
Figure 72-1
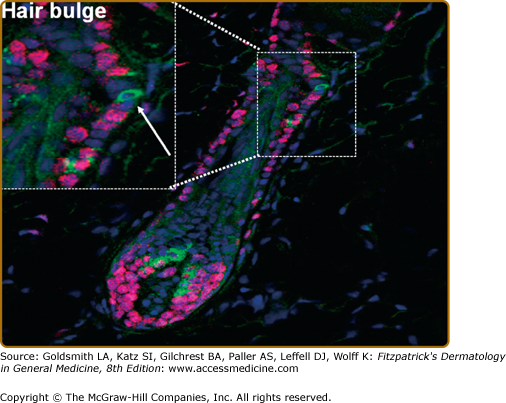
Melanocyte stem cells in the hair follicle bulge. A stem cell melanocyte is shown in the hair follicle bulge, indicated by an arrow in the high-power insert. These cells stain positive for TRP-2 (green fluorescence), an early marker of commitment to the melanocyte lineage, but are negative for the proliferation marker Ki-67 (red fluorescence) that characterizes melanocytes migrating down the follicle to the dermal papilla during the anagen phase of the hair cycle. (From Botchkareva NV et al: SCF/c-kit signaling is required for cyclic regeneration of the hair pigmentation unit. FASEB J 15:645, 2001, with permission.)
The largest number of melanocytes are present in the skin and hair follicles. While in most furred mammals melanocytes are found only in the hair follicle, in humans, melanocytes are also present in interfollicular epidermis, specifically in the basal layer.28 There is approximately one melanocyte per five or six basal keratinocytes. Melanocytes synthesize melanin, a pigmented polymer that is stored in cytosolic organelles called melanosomes that are transferred to keratinocytes through melanocyte dendritic processes (Fig. 72-2). As keratinocytes are continuously being desquamated, there is a constant need for synthesis and transfer of melanosomes from melanocytes to keratinocytes in order to maintain cutaneous pigmentation.
Figure 72-2
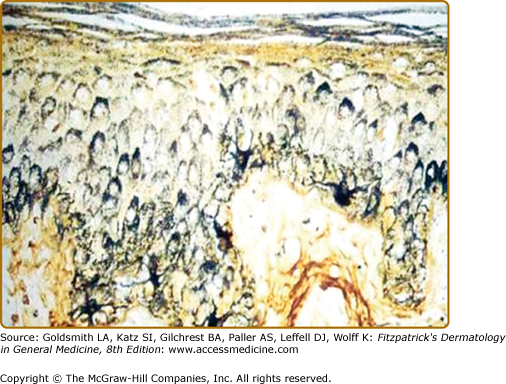
Melanosomes are organized into supranuclear “caps” within keratinocytes. Note melanized dendritic melanocytes and adjacent keratinocytes with the supranuclear “caps.” Melanin silver-stained (Fontana-Masson) section of a heavily melanized human epidermis. (Bar = 50 μm.) (From Byers HR et al: Role of cytoplasmic dynein in perinuclear aggregation of phagocytosed melanosomes and supranuclear melanin cap formation in human keratinocytes. J Invest Dermatol 121:813, 2003, with permission.)
The term “epidermal melanin unit” describes a single epidermal melanocyte surrounded by several epidermal keratinocytes31 (Fig. 72-3). Interestingly, signals from keratinocytes substantially regulate epidermal melanocyte survival, dendricity, melanogenesis and the expression of cell surface receptors32 (see below). Most keratinocyte-derived signals are induced by ultraviolet (UV) irradiation.
Figure 72-3
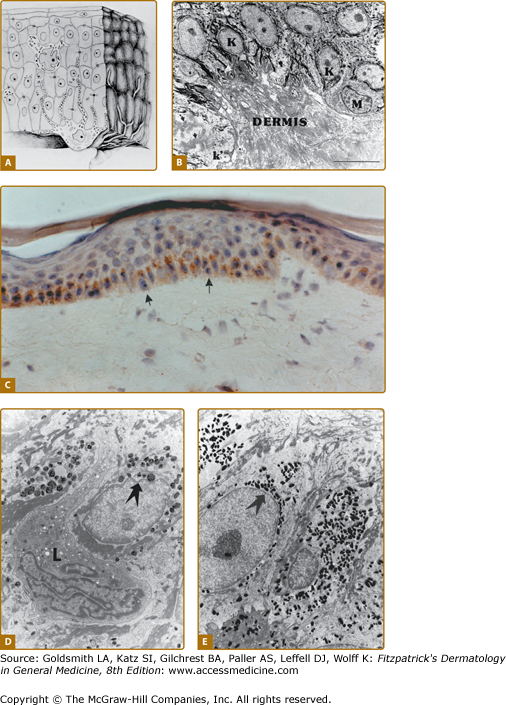
The epidermal melanin unit. A. Representation showing the relationship between basal melanocytes, keratinocytes, and Langerhans cells, shown at the upper layer of the epidermis. (From Quevedo WC Jr.: The control of color in mammals. Am Zoology 9:531, 1969, with permission.) B. Electron micrograph of the dermal–epidermal junction of human skin showing a dendritic melanocyte (M) among the basal keratinocytes (K). k′ represents a basal keratinocyte undergoing mitosis with condensed chromatin (arrows). (Bar = 10.0 μm.) (Illustration used with permission from Raymond E. Boissy, Department of Dermatology, University of Cincinnati, Cincinnati, Ohio.) C. Human epidermis immunostained for fibroblast growth factor-2 (FGF2). The figure shows basal keratinocytes with peroxidase reaction indicating the presence of immunore-active FGF2. Arrows point to melanocytes. (Used with permission from Glynis Scott, MD, Department of Dermatology, University of Rochester School of Medicine and Dentistry, Rochester, NY.) D and E. Distribution of melanosomes within keratinocytes in lightly pigmented Caucasian and darkly pigmented African-American skin. Melanosomes in lightly pigmented Caucasian skin (D) are distributed in membrane-bound clusters. In contrast, in darkly pigmented African-American skin (E) the melanosomes are individually distributed throughout the cytoplasm of epidermal keratinocytes. Melanosomes in both skin types are frequently concentrated over the apical pole of the nucleus (arrows). L = Langerhans cell. (Bar = 3.0 μm.) (Used with permission from Minwalla L et al: Keratinocytes play a role in regulating distribution patterns of recipient melanosomes in vitro. J Invest Dermatol 117:341, 2001.)
Melanocyte density/mm2 ranges from approximately 550 to >1,200 with the highest concentrations found in the genitalia and face.33,34 Melanocyte density is the same in individuals of different racial backgrounds,34 and thus cutaneous pigmentation does not depend on melanocyte number, but rather upon melanogenic activity within the melanocyte, the proportion of mature melanosomes, and/or their transfer and distribution within the keratinocytes.35 Indeed, in light-skinned individuals, melanosomes are smaller and are present in clusters within the keratinocytes, while in ethnic groups with darker complexion, melanosomes are larger, darker, and are individually dispersed within the keratinocytes.36
In contrast with interfollicular epidermal melanocytes, the follicular melanin unit undergoes cyclic modifications in coordination with the hair cycle (Fig. 72-4). Melanocytes are located in the proximal hair bulb during anagen and there is a ratio of 1:5 between melanocytes and keratinocytes and 1:1 between melanocytes and basal layer keratinocytes.37 Melanocytes proliferate, migrate, and undergo maturation during early to mid anagen. Melanogenesis and melanin transfer to keratinocytes occurs throughout anagen. Melanocytes eventually apoptose during late catagen. In mice, melanocyte proliferation and differentiation during anagen depends on c-Kit expression by melanocytes and SF synthesis by keratinocytes.38
Figure 72-4
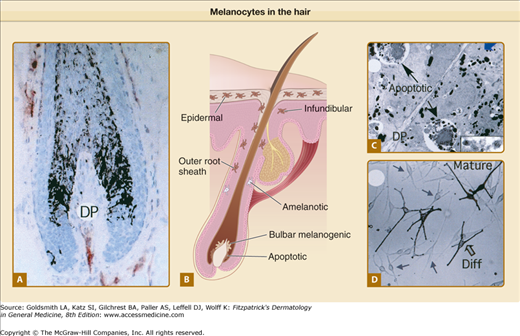
Melanocytes in the hair. A. Pigmented human scalp hair follicle in full anagen with high levels of hair bulb melanogenesis. Mature melanin granules are transferred into cortical keratinocyte. B. Scalp hair bulb. Representation of early catagen hair follicle showing loss of some bulbar melanotic melanocytes via apoptosis. Arrows point at melanocytes located in epidermal, infundibular, and outer root sheath regions. C. Transmission electron micrograph of section of an early catagen hair bulb showing apoptosis of melanotic melanocytes. Inset, high-power view of premelanosomes. D. Primary culture of human scalp hair follicle melanocytes. Mature, fully differentiated (Diff; large arrow), and less differentiated (small arrows) are indicated. DP = dermal papilla. (From Tobin DJ, Paus R: Graying: Gerontobiology of the hair follicle pigmentary unit. Exp Gerontol 36:29, 2001, with permission.)
Similar to their role in the epidermis, in hair, melanocytes transfer melanin to differentiated keratinocytes that ultimately form the hair shaft. They thus determine hair color by the amount of melanin transferred, as well as by the ratio of eumelanin (black–brown) to pheomelanin (red–yellow) (see below).39
In hair, melanin does not appear to have a protective effect, since UV irradiation does not reach the hair follicle. Still, in furry animals hair color plays an important role in camouflage, mimicry, and social communication.40 It is also speculated that melanocytes restrain keratinocyte proliferation, affect calcium homeostasis, and protect against reactive oxygen species (ROS) during the rapid proliferation and differentiation of the hair follicle.37
 Ocular melanocytes are found in the uveal tract.28 Unlike cutaneous melanocytes, ocular melanocytes are in contact only with each other and they do not transfer their melanosomes (eFig. 72-4.1). It is proposed that ocular melanocytes are exposed to high oxygen tension and melanin serves as protection against oxidative damage. Iris melanin may be required to protect capillaries, muscles, and motor nerves that control pupil contraction.28
Ocular melanocytes are found in the uveal tract.28 Unlike cutaneous melanocytes, ocular melanocytes are in contact only with each other and they do not transfer their melanosomes (eFig. 72-4.1). It is proposed that ocular melanocytes are exposed to high oxygen tension and melanin serves as protection against oxidative damage. Iris melanin may be required to protect capillaries, muscles, and motor nerves that control pupil contraction.28
eFigure 72-4.1

Ocular tissue demonstrating the choroid (C), the retinal pigment epithelium (R), and the photoreceptor cells of the retina (P). The connective tissue of the choroid contains numerous fibroblasts (F) and dendritic melanocytes (M) with cytoplasm filled with mature melanosomes. The retinal pigment epithelium is a single layer of polarized cuboidal melanocytes containing relatively elongated and mature melanosomes both within the cell body of the retinal pigment epithelium cells and in the numerous slender apical processes (arrows) that interdigitate between the rod and cone photoreceptor cells. (Bar = 10.0 μm.) (Illustration used with permission from Raymond E. Boissy, Department of Dermatology, University of Cincinnati, Cincinnati, Ohio, USA.)
 Melanocytes appear to be essential for the development and function of the eye and optic nerve.41 It is thought that they may play a role in the proper routing of ipsilateral and contralateral neural fibers in the optic chiasm during early development of the optic cup,42,43 as in the absence of melanin, the path of the highly intermixed optic nerve axons is deranged, resulting in disruption of the topographic relationship of the nerve fibers.44,45 The importance of melanin for proper ocular function is demonstrated by the visual abnormalities observed in patients with albinism.44
Melanocytes appear to be essential for the development and function of the eye and optic nerve.41 It is thought that they may play a role in the proper routing of ipsilateral and contralateral neural fibers in the optic chiasm during early development of the optic cup,42,43 as in the absence of melanin, the path of the highly intermixed optic nerve axons is deranged, resulting in disruption of the topographic relationship of the nerve fibers.44,45 The importance of melanin for proper ocular function is demonstrated by the visual abnormalities observed in patients with albinism.44
 Melanoctyes reside in the cochlea28 and are important for hearing, as loss of otic melanocytes leads to deafness. It is thought that otic melanocytes help in the maintenance of the endolymph through the regulation of potassium transport.46 The endolymphatic fluid in the cochlea is one of the few extracellular fluids with high concentrations of positively charged potassium, and melanocytes are believed to play a critical role in its maintenance, probably by transporting potassium from areas of high concentration to areas of low concentration through plasma membrane ionic channels.47 In the absence of cochlear melanocytes, the endolymphatic potential is low, leading to deafness.48
Melanoctyes reside in the cochlea28 and are important for hearing, as loss of otic melanocytes leads to deafness. It is thought that otic melanocytes help in the maintenance of the endolymph through the regulation of potassium transport.46 The endolymphatic fluid in the cochlea is one of the few extracellular fluids with high concentrations of positively charged potassium, and melanocytes are believed to play a critical role in its maintenance, probably by transporting potassium from areas of high concentration to areas of low concentration through plasma membrane ionic channels.47 In the absence of cochlear melanocytes, the endolymphatic potential is low, leading to deafness.48
 Loss of melanocytes at any age can eventually lead to deafness, and patients with Waardenburg syndrome type 2 (see Chapter 72) are deaf because of lack of melanocytes in the inner ear.49,50 Interestingly, the preservation of hearing in albinos indicates that melanin production within otherwise viable melanocytes is not essential for hearing. However, it appears that since albinos are more susceptible than the normal population to hearing loss from noise and/or exposure to toxic agents,51,52 melanin must provide some protective effect.
Loss of melanocytes at any age can eventually lead to deafness, and patients with Waardenburg syndrome type 2 (see Chapter 72) are deaf because of lack of melanocytes in the inner ear.49,50 Interestingly, the preservation of hearing in albinos indicates that melanin production within otherwise viable melanocytes is not essential for hearing. However, it appears that since albinos are more susceptible than the normal population to hearing loss from noise and/or exposure to toxic agents,51,52 melanin must provide some protective effect.

Melanization
The major differentiated function of melanocytes is to synthesize melanin in specialized organelles within the melanocytes, the melanosomes, and to transfer melanosomes to neighboring keratinocytes in order to provide protection from UV irradiation (Fig. 72-5). Melanogenesis, the synthesis and distribution of melanin in the epidermis, involves several steps: transcription of proteins required for melanogenesis; melanosome biogenesis; sorting of melanogenic proteins into the melanosomes; transport of melanosomes to the tips of melanocyte dendrites and transfer of melanosomes to keratinocytes. Disruption in any of these events results in hypopigmentation.
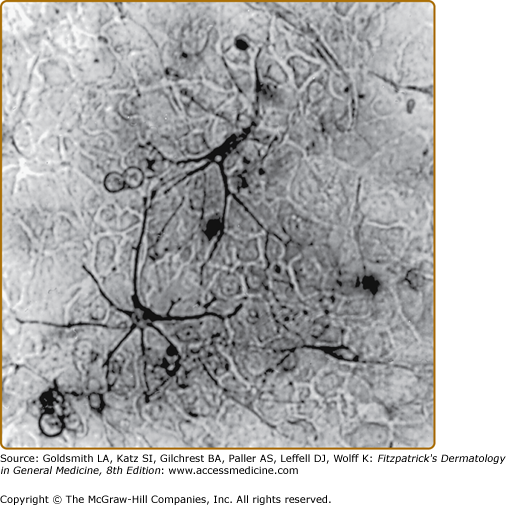
Melanosomes
The melanosome is a unique membrane-bound organelle in which melanin biosynthesis takes place. Because melanosomes contain enzymes and other proteins also present in lysosomes, they are thought to represent a modified version of the latter. Proteins common to both organelles include the lysosome-associated membrane proteins (LAMPs) that participate in autophagy and regulation of intravesicular pH,56 as well as acid phophatase, a marker enzyme for lysosomes.56 Also like lysosomes, melanosomes can endocytose receptors that are targeted for degradation.56
Depending on the type of melanin synthesized, melanosomes can be divided into eumelanosomes and pheomelanosomes (Fig. 72-6). Eumelanosomes are large (∼0.9 × 0.3 mm), elliptical in shape and contain a highly structured fibrillar glycoprotein matrix required for eumelanin synthesis.40 Pheomelanosomes are smaller (∼0.7 mm in diameter), spherical in shape and their glycoprotein matrix appears disorganized and loose40 Although both eumelanosomes and pheomelanosomes may be present within a single melanocyte,57 once committed, they do not change.58
Figure 72-6
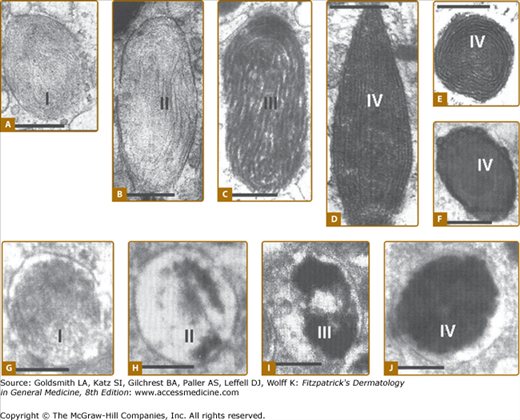
Melanosome biogenesis. Electron microscopy of eumelanosome (A–F) and of pheomelanosome (G–J) development. I, II, III, and IV in A–J represent the different maturation stages of melanosomes. [Scale bars are as follows (in μm): a = 0.20; b = 0.23; c = 0.24; d = 0.22; e = 0.20; f = 0.35; h = 0.26; i = 0.26; j = 0.30; k = 0.5.] (From Slominski A et al: Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev 84:1155, 2004, with permission from the American Physiological Society.)
Melanosomes display four maturation stages (Fig. 72-6). Stage I melanosomes or premelanosomes likely develop from the endoplasmic reticulum (ER).40 They have an amorphous matrix and display internal vesicles that form as a result of membrane invagination. Premelanosomes already contain the glycoprotein Pmel17 (gp100) but it requires further processing to become a component of the final fibrillar matrix.59 Stage II eumelanosomes have organized structured fibrillar matrix, but no active melanin synthesis, whereas in stage II pheomelanosomes melanin synthesis already takes place. Although no active melanogenesis takes place in stage II eumelanosomes, they already contain the enzyme tyrosinase. Deposition of melanin on the fibrillar matrix is found in stage III eumelanosomes, while stage IV eumelanosomes are fully melanized and their internal matrix is masked by melanin deposits.60,61
The timely and organized sorting of melanogenic enzymes and structural proteins to melanosomes is an integral part of melanosomal maturation. Melanosome proteins express sorting signals at their amino-terminus and these direct them into the ER and eventually into the melanosomes.40,60,61
Tyrosinase is present in plants, insects, amphibians, and mammals. It was initially identified in the early 1900s in mushroom extracts and was subsequently isolated and purified in 1949 from murine melanoma cells.62 Mouse and human tyrosinase genes are 60–70 kb, and 50 kb long, respectively. The murine tyrosinase gene maps to chromosome 7, whereas human tyrosinase gene maps to chromosome 11. The human tyrosinase gene is composed of five exons and four introns,63 and tyrosinase mRNA is approximately 2 kb long (genebank access number NM_000372).
Tyrosinase is synthesized in the ER as a precursor protein whose nascent chain is processed in the Golgi complex where sialic acid and neutral sugars are added to the peptide via N– and O-glycosidic linkages through a process called glycosylation64 (Fig. 72-7). At least four forms of tyrosinase, all differing with regards to their degree of glycosylation, have been identified. The glycolsylation steps have been shown to be important for proper association of tyrosinase with melanosomes, as well as for its activity.64 Following the glycosylation steps, mature tyrosinase is folded in the ER, a step required for appropriate trafficking/sorting of tyrosinase into the Golgi apparatus and ultimately into endosomes and finally into melanosomes. A strict control mechanism guarantees the elimination of defective tyrosinase.
Figure 72-7
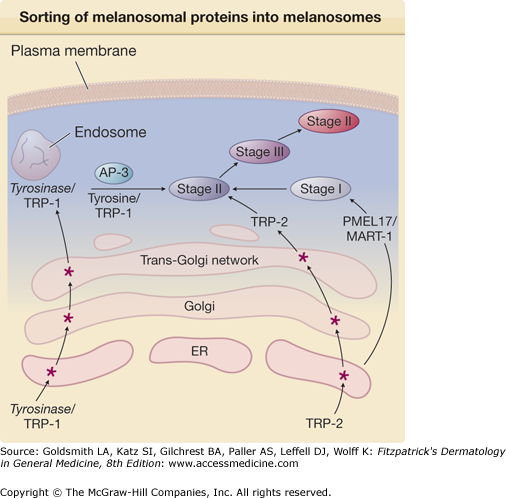
Sorting of melanosomal proteins into melanosomes. Tyrosinase and tyrosinase-related protein-1 (TRP-1) are initially synthesized in the endoplasmic reticulum (ER) and, after additional maturation steps (*) in the Golgi and trans-Golgi network, are packaged in endosomes as a complex. TRP-2 follows similar maturation steps. Melanosomes originate in the ER as stage I already containing the melanosomal proteins PMEL17 and MART-1. They then mature to stage II melanosomes and fuse with tyrosinase/TRP-1 in a process directed by the adaptor protein 3 (AP-3). Melanosomes become progressively darker as melanin biosynthesis takes place.
Within the melanosome, tyrosinase spans melanosomal outer membrane (eFig. 72-7.1). It has three domains: (1) an inner melanosomal domain, (2) a melanosomal transmembrane domain, (3) and a cytoplasmic domain. The inner domain that contains the catalytic region is approximately 90% of the protein. It is followed by a short transmembrane domain, and a cytoplasmic domain composed of approximately 30 amino acids.65 Histidine residues present in the inner (catalytic) portion of tyrosinase bind copper (Cu) ions and the latter are required for tyrosinase activity.
eFigure 72-7.1
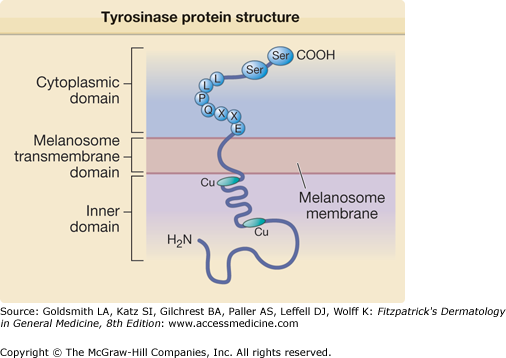
Schematic diagram of tyrosinase protein structure. Tyrosinase protein can be divided into three domains: the inner melanosomal domain, the transmembrane domain, and the cytoplasmic domain. The inner domain contains the catalytic activity of the enzyme and the copper (Cu)-binding sites. The cytoplasmic domain contains the sequence (EXXQPLL) that participates in cellular trafficking of tyrosinase into melanosomes. The cytoplasmic domain also participates in regulating tyrosinase activity through phosphorylation of serine residues (Ser).
The biological function of the tyrosinase cytoplasmic domain was not known for a long time. In a mouse model where the entire cytoplasmic domain is missing, tyrosinase protein is inserted into the cellular plasma membrane instead of into the melanosomal membrane, suggesting that tyrosinase cytoplasmic domain is required for proper trafficking of tyrosinase into melanosomes. Indeed, it was found that the motif EXXQPLL (glutamic acid-X-X-glutamine-proline-leucine-leucine, where “X” stands for any amino acid) in the cytoplasmic domain is responsible for tyrosinase trafficking into the melanosomes.66 In addition, protein kinase C-β (PKC-β) (see below) must phosphorylate two serine residues on this cytoplasmic domain to activate tyrosinase,65 and in the absence of those phosphorylation events pigmentation does not occur.
Tyrosinase mutations including missense, nonsense frameshift, and deletion mutations that lead to inactivation of the enzyme are found in oculocutaneous albinism type I (see Chapter 73 and Albinism database: http://albinismdb.med.umn.edu/). Such mutations may affect tyrosinase glycosylation interfering with enzyme maturation, or may involve Cu-binding sites disrupting tyrosinase activity or premature termination of tyrosinase protein that causes truncation of cytoplasmic domain.64
Two tyrosinase-related proteins, (1) TRP-1 and (2) TRP-2, play important roles in melanogenesis.67–69 They are structurally related to tyrosinase and share ∼40% amino acid homology. Also, similar to tyrosinase, TRP-1 and TRP-2 are glycoproteins located within the melanosomes and span the melanosomal membrane.70 The conserved nucleotide and amino acid sequences among these three melanogenic enzymes suggest that they originated from a common ancestral gene.71,72 Mutations of TRP-1 and TRP-2 in mice after coat color (“brown” and “slaty” mice, respectively) and polymorphisms of these gene products are implicated in lighter hair and skin color in European population studies. Their functions are incompletely understood, but the proteins complex with tyrosinase and may stablilize it.
 In mice, TRP-1 maps to the brown locus on chromosome 4 and spans ∼18 kb on the genomic DNA.69,72 The human homolog of TRP-1 maps to chromosome 9 and spreads over 24 kb of the genomic DNA.72 Like tyrosinase, TRP-1 is synthesized in the ER and undergoes several glycosylation steps (Fig. 72-7). The exact function of TRP-1 in melanin biosynthesis, especially in human melanogenesis, is not well understood. In mice, TRP-1 displays a wide range of enzymatic activities including DOPA,73 tyrosine hydroxylase,74 DOPA oxidase,69,75 catalase,76 and DHICA (5,6-dihydroxyindole-2-carboxylic acid) oxidase67,77 (see Section “Melanin Biosynthesis”). Conversely, in humans TRP-1 does not display DHICA oxidase activity and the additional activities described above have not been reported to date for this enzyme.
In mice, TRP-1 maps to the brown locus on chromosome 4 and spans ∼18 kb on the genomic DNA.69,72 The human homolog of TRP-1 maps to chromosome 9 and spreads over 24 kb of the genomic DNA.72 Like tyrosinase, TRP-1 is synthesized in the ER and undergoes several glycosylation steps (Fig. 72-7). The exact function of TRP-1 in melanin biosynthesis, especially in human melanogenesis, is not well understood. In mice, TRP-1 displays a wide range of enzymatic activities including DOPA,73 tyrosine hydroxylase,74 DOPA oxidase,69,75 catalase,76 and DHICA (5,6-dihydroxyindole-2-carboxylic acid) oxidase67,77 (see Section “Melanin Biosynthesis”). Conversely, in humans TRP-1 does not display DHICA oxidase activity and the additional activities described above have not been reported to date for this enzyme.
 Nevertheless, the presence of TRP-1 appears to be required for melanin synthesis in human melanocytes, as TRP-1 absence results in the pigmentary disorder oculocutaneous albinism type 378 (see Chapter 72). Since recently TRP-1 was shown to influence tyrosinase activity by forming a complex with tyrosinase, it is possible that TRP-1 plays a role in tyrosinase activation and/or stabilization.79,80 A known TRP-1 function in both murine and human melanocytes at least in vitro is to increase the ratio of eumelanin to pheomelanin.81,82 TRP-1 may also play a role in melanosomal biogenesis, as suppression of TRP-1 is associated with structurally abnormal melanosomes.61
Nevertheless, the presence of TRP-1 appears to be required for melanin synthesis in human melanocytes, as TRP-1 absence results in the pigmentary disorder oculocutaneous albinism type 378 (see Chapter 72). Since recently TRP-1 was shown to influence tyrosinase activity by forming a complex with tyrosinase, it is possible that TRP-1 plays a role in tyrosinase activation and/or stabilization.79,80 A known TRP-1 function in both murine and human melanocytes at least in vitro is to increase the ratio of eumelanin to pheomelanin.81,82 TRP-1 may also play a role in melanosomal biogenesis, as suppression of TRP-1 is associated with structurally abnormal melanosomes.61
 In mice, TRP-2, also known as DOPA, maps to the Slaty locus on chromosome 14, whereas human TRP-2 maps to chromosome 13.68 Mouse and human TRP-2 share 84% nucleotide homology.83 Like tyrosinase and TRP-1, TRP-2 is a glycoprotein84 that is synthesized in the ER and undergoes several maturation steps in the Golgi and trans-Golgi network (Fig. 72-7). Like tyrosinase and TRP-1, it eventually localizes to melanosomes and it spans the melanosomal membrane. Within the melanosome, TRP-2 is complexed with tyrosinase and TRP-1. During melanin synthesis, TRP-2 converts DOPAchrome to the carboxylated derivative DHICA. As in the case for tyrosinase, TRP-2 also requires metal ions for its enzymatic activity, but it is zinc rather than copper (Cu) that is required for TRP-2 function.85
In mice, TRP-2, also known as DOPA, maps to the Slaty locus on chromosome 14, whereas human TRP-2 maps to chromosome 13.68 Mouse and human TRP-2 share 84% nucleotide homology.83 Like tyrosinase and TRP-1, TRP-2 is a glycoprotein84 that is synthesized in the ER and undergoes several maturation steps in the Golgi and trans-Golgi network (Fig. 72-7). Like tyrosinase and TRP-1, it eventually localizes to melanosomes and it spans the melanosomal membrane. Within the melanosome, TRP-2 is complexed with tyrosinase and TRP-1. During melanin synthesis, TRP-2 converts DOPAchrome to the carboxylated derivative DHICA. As in the case for tyrosinase, TRP-2 also requires metal ions for its enzymatic activity, but it is zinc rather than copper (Cu) that is required for TRP-2 function.85
PKC constitutes a family of at least 12 isoforms86 among that PKC-β has been shown to be involved in regulating tyrosinase activity.87 The mechanisms through which PKC mediates a wide range of membrane generated signals and their relevance to melanocyte biology are further discussed below (see Section “Signaling Pathways Regulating Melanocyte Function”). PKC-β phosphorylates serine residues on the cytoplasmic domain of tyrosinase, thus activating tyrosinase.88 Still, the means by which PKC-β-mediated phosphorylation of tyrosinase leads to the enzyme activation is not well elucidated. It has been suggested that phosphorylation of tyrosinase causes a complex to form between tyrosinase and TRP-1,89 an event known to stabilize tyrosinase and increase its enzymatic activity.79
In melanocytes, activated PKC-β is associated with melanosomes and the enzyme is found in close proximity to the melanosomal membrane.88 While structural differences among PKC isoforms may contribute to their associations with particular subcellular fractions, receptors for activated C-kinase (RACK), unique for each PKC isoform, primarily determine the translocation of specific PKC isoforms to specific cellular compartments to activate its target on the membrane90–92 (Fig. 72-8). RACK-I is the PKC-β partner,90 and in human melanocytes, the activated PKC-β/RACK-I complex translocates to the melanosome membrane to allow tyrosinase phosphorylation (Fig. 72-8).93
Figure 72-8
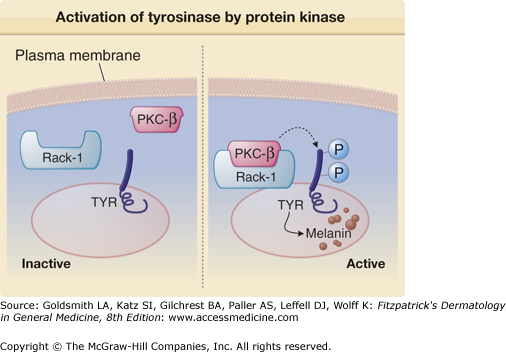
Activation of tyrosinase by protein kinase C-β (PKC-β). Under baseline conditions, there is no activation of PKC-β, and tyrosinase (TYR) is not phosphorylated. Activated PKC-β binds receptors for activated C-kinase-I (RACK-I), the complex translocates to the melanosome, and phosphorylates serine residues on the cytoplasmic tail of tyrosinase. Tyrosinase phosphorylation activates the enzyme to catalyze melanin biosynthesis.
Fibrillar matrix proteins within the melanosomes are required for proper deposition of melanin. Pmel17 and MART-1 are two such melanosomal structural matrix proteins.
Pmel17, also known as gp100 and the silver locus product, is a glycoprotein recognized by the antibodies HMB45, HMB50, and NKI-β.94 It plays a critical role in fibril matrix formation within eumelanosomes.59,95 Pmel17 transcription is induced by α-MSH through MITF and it is synthesized as a precursor protein in the ER and the protein undergoes glycosylation and eventual cleavage (Fig. 72-7). After its synthesis, Pmel17 is transported to stage I melanosomes to form a fibrillar structure that is the backbone of eumelanosome matrix,94 contributes to melanosome ellipsoid shape and promotes melanin polymerization.94 Melanosomes lacking Pmel17 cannot transit to stage II and have no active melanogenesis.96 It has been suggested that loss of functional Pmel17 results in melanin cytotoxicity, perhaps through leakage of melanin intermediates from abnormal melanosomes into the cytosol.94,97 However, there are no known hypopigmentary disorders in humans linked to mutations of Pmel17.
MART-1, also known as Melan A, is a membrane-associated protein98 that is present in stage I and stage II melanosomes and forms a complex with Pmel17 (Fig. 72-7). MART-1 affects the expression, stability, trafficking, and processing of Pmel17 within the melanosomes.98 To date, no hypopigmented phenotypes associated with nonfunctional MART-1 have been identified.
Sorting of membrane-associated proteins including tyrosinase, TRP-1, TRP-2, and Pmel17 and directing them to the appropriate cytosolic organelles is facilitated by heterodimeric adapter protein complexes (APs).99,100 AP-3 and possibly also AP-1 facilitate tyrosinase transport from endosomes to melanosomes101 (Fig. 72-7). Patients with Hermansky–Pudlak syndrome—an autosomal recessive disorder of oculocutaneous albinism, platelet dysfunction, and pulmonary disease (see Chapter 73)—have defects in specific subunits of the AP-3 adaptor protein complex and as a result display several anomalies associated with cellular transport of molecules.102 Studies suggest that a molecule of the kinesin family, microtubule-associated motor proteins, is involved in endosomal sorting and positioning of melanosomal proteins via interaction with AP-1.103,104
The P-protein (pink-eyed dilution) is a transmembrane protein with 12 membrane-spanning domains whose sequence is homologous to that of other transmembrane transport proteins, including anion transporters97,105,106 thought to function as a transport protein.107 Studies have identified the protein as an ATP-associated proton pump responsible for maintaining acidic environment within the melanosomes.108 Other proposed functions of P-protein include stabilizing the tyrosinase/TRP-1/TRP-2 complex and/or transporting tyrosine into the melanosomes.105 Individuals lacking functional P-protein display occulocutaneous albinism type 2, largely due to improper melanosomal pH.108–110 Also, Angelman and Prader–Willi syndromes display deletion mutations that include the P-locus on chromosome 15.
SLC24A5 is a melanosomal protein whose structure and homology to cation exchange proteins suggests that it is a melanosome-associated cation exchanger.111 Mutations in slc24a5 in zebrafish lead to hypopigmentation of the organism.111 The ancestral human homolog is expressed by darker complexioned individuals including Africans and Asians, while lighter complexioned Europeans tend to express a variant allele.111
LAMPs are linked to melanosome membranes and/or matrix. They are thought to protect melanosomal integrity by acting as scavengers of free radicals that are produced during melanin biosynthesis.112 Because LAMPs are also present in lysosomes, it is thought that melanosomes and lysosomes share a common ancestral origin.112
MITF, a basic-helix-loop-helix (bHLH) and leucine zipper transcription factor, has been termed the master gene for melanocyte survival and is a key factor regulating the transcription of the major melanogenic proteins, tyrosinase, TRP-1, TRP-2,12 PKC-β113 and MART-1.114 In melanocytes, it is the MITF-M isoform that stimulates transcription of tyrosinase and PKC-β.114 MITF binds to conserved consensus elements in gene promoters, specifically the M- (AGTCATGTGCT) and E- (CATGTG) boxes.115 It can bind as a homodimer or a heterodimer with another related family member.116 MITF appears to be a key regulator determining cell fate, as transfection of human MITF cDNA into mouse fibroblasts converts these cells into dendritic cells expressing melanocyte-specific genes.117
MITF comprises a family of nine isoforms: (1) MITF-M, (2) -A, (3) -B, (4) -H, (5) -C, (6) -D, (7) -E, (8) -J, and (9) -Mc.118,119 MITF-M expression is highly specific for melanocytic cells.120 Melanocytes express in addition other MITF isoforms specifically, MITF-A, -B, and, -E.114 The biologic role of other MITF isoforms in normal melanocytes is not known.
The activity and stability of MITF are modulated by phosphorylation of the protein. MITF activity is increased upon its phosphorylation by the mitogen-activated protein kinase-2 (MAP kinase-2), whose activity is in turn induced by binding of SF/kit/stem cell factor to c-Kit receptor121 (Fig. 72-8). Phosphorylated MITF binds to another protein, p300/CBP, which belongs to a coactivator family of proteins and acts to enhance MITF transcriptional activity.121,122 Another kinase that is activated by SF/c-Kit interaction is p90RSK that also phosphorylates MITF but at a different site from that phosphorylated by MAP kinase-2.123 These phosphorylation events both activate MITF and at the same time decrease the stability of the protein, as phosphorylated MITF is targeted for degradation by proteosomes (eFig. 72-8.1A).123,124
eFigure 72-8.1
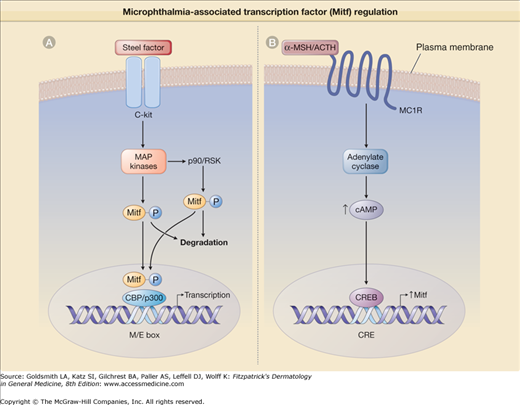
Microphthalmia-associated transcription factor (MITF) regulation. A. MITF is activated when steel factor (SF) binds to its cell surface receptor c-Kit. SF–c-Kit interaction activates mitogen-activated protein (MAP) kinases, which then phosphorylate MITF. Phosphorylated MITF then recruits the cotranscription activator CBP/p300, and the complex binds to the M- or E-boxes (M/E box) within the promoter of target genes to regulate their transcription. MITF can also be phosphorylated at a different site by p90/RSK kinase. Both phosphorylation events lead to MITF ubiquitination and subsequent proteosome-mediated degradation. B. On binding to melanocortin receptor-1 (MC1R), α-melanocyte-stimulating hormone (α-MSH)/adrenocorticotropic hormone (ACTH) activate the enzyme adenylate cyclase that upregulates the cyclic adenosine monophosphate (cAMP) level, leading to cAMP-response element binding protein (CREB) activation and binding its DNA consensus sequence CRE in the MITF promoter to induce MITF transcription.
The expression of MITF is under the control of several transcription factors, including Sox10 (mutated in Waardenburg’s syndrome type 4, see Chapter 73) and Pax3. MITF expression is also controlled by the cAMP-response element binding protein (CREB) and Lef1 transcription factor that participates in Wnt-signaling. These transcription factors bind to specific sites within MITF promoter regions to induce MITF transcription.116 The promoter region of the MITF gene contains a cAMP-response element (CRE) that interacts with CREB when the cAMP-dependent pathway is activated.125,126 Therefore, cAMP-elevating agents like α-MSH induce the expression of MITF (eFig. 72-8.1B).
MITF promotes melanoycte survival by upregulating the expression of a major antiapoptotic protein BCl2.127 It is frequently overexpressed or amplified in melanomas, contributing to their increased survival.128–131 Mutations in MITF are found in the pigmentary disorder Waardenburg syndrome type 2132(see Chapter 73).
A role for MITF in melanocyte proliferation has also been proposed, as under certain conditions, MITF induces the expression of the cell cycle-associated kinase Cdk2 that is involved in the progression of cells from G1 into S phase of the cell cycle.133 MITF also suppresses the expression of p21, a protein that inhibits Cdk2 activation.133,134 Conversely, under different conditions, MITF can induce p21 expression and it can also stimulate the expression of p16INK4a, a protein that inhibits the activation of kinases required for progression through the cell cycle, thus promoting cell cycle arrest.135,136 Because MITF cooperates with other transcription factors to induce its effects, it is to be expected that these transcription factors would influence MITF activity, resulting in either stimulation or inhibition of melanocyte proliferation.
Mice bearing null mutations of MITF display loss of melanocytes, deafness and failure of differentiation of retinal pigment epithelium.12
Melanocortin receptors (MCRs) comprise a family of five related receptors (MC1R, MC2R, MC3R, MC4R, and MC5R). Each has seven transmembrane domains and they belong to the G-protein-coupled receptor superfamily.137 MC3R and MC4R are mainly found in the central nervous system, are absent in melanocytes,138 and are thought to control energy intake. MC2R is expressed in the adrenal cortex and MC5R is expressed in peripheral adipocytes.139 MC1R is expressed in a number of cells such as endothelial cells, fibroblasts,140 and keratinocytes,140 but the highest expression is found in melanocytes.140
α-MSH and adrenocorticotropic hormone (ACTH), a 39 amino acid proopiomelanocortin-derived peptide that contains the α-MSH sequence (Fig. 72-9A),141–143 activate MC1R (Fig. 72-9B) Receptor–ligand interaction leads to G-protein-dependent activation of the enzyme adenylate cyclase followed by increased intracellular cAMP level141 inducing MITF transcription and upregulating the level of melanogenic proteins including tyrosinase40 promoting the synthesis of brown/black eumelanin.141 Agouti, a protein expressed in both humans and mice, whose expression in mice leads to yellow coat color, antagonizes α-MSH by competitive binding to MC1R. It thus blocks adenylate cyclase activation144–146 and favors pheomelanin over eumelanin synthesis (Fig. 72-10). However, the role of agouti in human pigmentation is poorly documented.
Figure 72-9
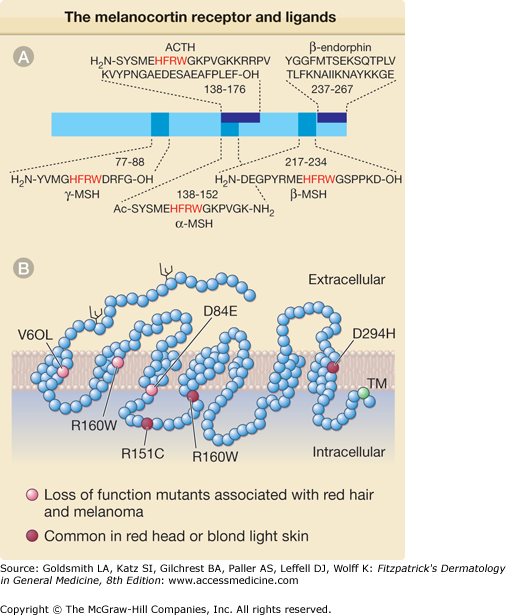
The melanocortin receptor (MCR) and its ligands. A. Structure of the proopiomelanocortin precursor. Standard abbreviations for amino acids are used. The synthetic superactive α-melanocyte-stimulating hormone (α-MSH) analogue [Nle4 D-Phe7]-α-MSH is modified by the exchange of methionine (M) with norleucine and L-phenylalanine (F) with D-phenylalanine. In red are critical amino acids required for binding to the MCR. B. Schematic representation of the human MC1R receptor. Each of the 318 amino acid residues in the polypeptide chain of the receptor is represented by an empty circle. Branched structures represent N-linked glycosylation sites. Reduced function mutants (red circles), variants common in red- or blond-haired and fair skinned individuals (orange circles), and the conserved C-terminal cysteine (green circle), the possible site for fatty acid acylation and anchoring to the plasma membrane, are indicated. Ac = acetylated; ACTH = adrenocorticotropic hormone; NH2 = amidated; TM = transmembrane domain.
Figure 72-10
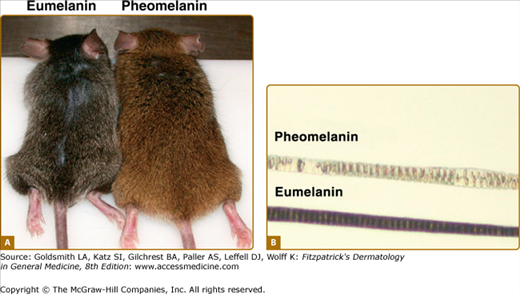
Eumelanin and pheomelanin presentation in mice. A. Two mice with different coat colors are shown. The one on the left displays brown/black coat color due to eumelanin, and the one on the right displays red/yellow coat color due to pheomelanin. B. Representative hair shafts of these mice. (From Sharov et al: Bone morphogenic protein (BMP) signaling controls hair pigmentation by means of cross-talk with the melanocortin receptor 1 pathway. PNAS 102:93, 2004, with permission.)
Polymorphisms within the MC1R gene are largely responsible for the different skin/hair color among different racial groups.147 At least 30 MC1R variants have been identified and nine of them display loss of function148,149 (Fig. 72-9B), not being able to induce intracellular cAMP production in response to α-MSH despite adequate receptor/ligand binding. Other MC1R variants have reduced affinity for α-MSH.148,149 Three MC1R variants, each with only a single amino acid substitution, have been associated with red/yellow hair and fair skin150 of Northern Europeans and Australians.151–156 Mice expressing a loss-of-function MC1R variant receptor also fail to respond to UV irradiation with increased pigmentation despite an increased level of epidermal α-MSH,157 but do tan if provided forskolin, a chemical enhancer of pigmentation that bypasses the receptor to directly increase cAMP, demonstrating that the intracellular melanogenic pathway is functional in such individuals.155
Melanin Biosynthesis
Two types of melanins are synthesized within melanosomes: (1) eumelanin and (2) pheomelanin.158 Eumelanin is dark, brown–black, and insoluble, whereas pheomelanin is light, red–yellow sulfur-containing, and soluble.158 Melanins are indole derivatives of 3,4 di-hydroxy-phenylalanine (DOPA) and they are formed in melanosomes through a series of oxidative steps159 (Fig. 72-11). Melanosomal pH affects the activity of the melanogenic enzymes and influences melanin polymerization.
Figure 72-11
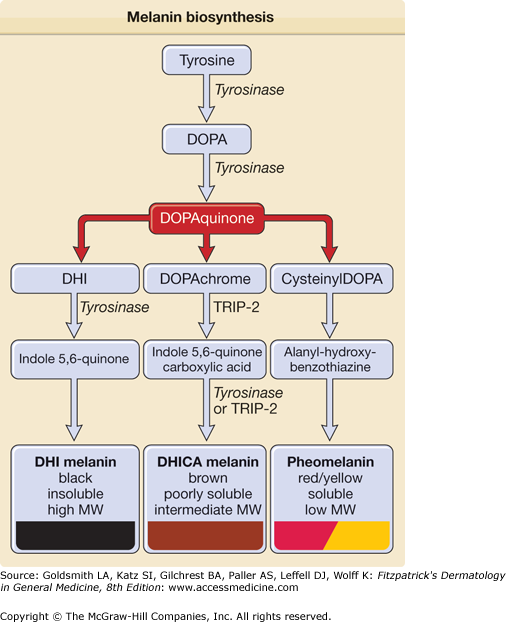
Melanin biosynthesis. Melanin biosynthesis begins with the amino acid tyrosine that is converted to DOPA (3,4-dihydroxyphenylalanine) in the rate-limiting step of melanin biosynthesis catalyzed by tyrosinase. DOPA is subsequently converted to DOPAquinone by the same enzyme. DHI (5,6-dihydroxyindole) and DHICA (5,6-dihydroxyindole-2-carboxylic acid) are then formed to produce either black or brown eumelanin. Alternatively, through incorporation of glutathione or cysteine, DOPAquinone can form pheomelanin. MW = molecular weight; TRP = tyrosinase-related protein.
The synthesis of both types of melanin involves the rate-limiting catalytic step in which the amino acid tyrosine is oxidized by the enzyme tyrosinase (also called tyrosine oxidase, DOPA oxidase, monophenol, DOPA: oxygen oxidoreductase) to DOPA, a first step in a reaction known as the Raper–Mason pathway160 (Fig. 72-11). Conversion of tyrosine to DOPA is thought to be the critical rate-limiting step in melanogenesis, as inhibition of this reaction blocks melanin synthesis.161 In both reactions, DOPA acts as a cofactor and also as a substrate for tyrosinase. Although the exact interaction between tyrosinase and its substrates is not completely understood, in vitro kinetic studies suggest that distinct sites mediate tyrosinase binding to tyrosine and to DOPA, and that binding to DOPA causes a conformational change in tyrosinase, resulting in increased affinity for both tyrosine and DOPA.
DOPA is oxidized into DOPAquinone,162 DOPAquinone is further converted to DOPAchrome and DOPAchrome can be converted to 5,6-dihydroxyindole (DHI) or to 5,6-dihydroxyindole-2-carboxylic acid (DHICA). The latter reaction is catalyzed by the enzyme DOPAchrome tautomeras or TRP-2. The level of brown versus black eumelanin appears to correlate with DHI/DHICA ratio, with a higher ratio leading to the formation of black eumelanin and a lower ratio to brown eumelanin.163 DOPAquinone can also combine with glutathione or cyteine to form cysteinylDOPA, which then becomes the yellow/red, soluble, low-molecular-weight pheomelanin.163 Interestingly, tyrosinase also catalyzes a more distant step in melanin biosynthesis, namely DHI conversion to indole-5,6-quinone. In mice, the enzyme TRP-1 (also called DHICA oxidase) converts DHICA to indole-5,6-quinone carboxylic acid. However, the TRP-1 role in human melanin biosynthesis is not well established.
The main function of melanin is to provide protection against UV-induced DNA damage by absorbing and scattering UV radiation (280–400 nm). Accordingly, energy absorption by melanin is maximal in this portion of the electromagnetic spectrum, and decreases gradually across the visible light spectrum. UV absorbed by melanin is converted into heat, a less toxic form of energy.164 Still, in vitro studies conducted by several investigators suggest that melanin’s capacity to act as a sunscreen is limited and that melanin, when incorporated into a cream and spread over the skin, absorbs only 50%–75% of incident sunlight. Naturally, it is possible that in vivo, by virtue of localizing above the nucleus, melanin in melanosomes achieves a higher level of protection.
Melanin intermediates as well as melanin itself can also be harmful to the cell because, depending on their molecular weight and polymerization state, they can promote UVA (320–400 nm)-induced DNA damage, most likely through the generation of ROS.165 It has been suggested that the increased incidence of UV-induced melanomas in light-skinned, red-hair individuals is not only due to decreased ability of pheomelanin to protect against UV-induced DNA damage, but may also be due to mutagenic capacity of pheomelanin and possibly other melanin intermediates as a result of their pro-oxidant capacity.166
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


