Fig. 12.1
The spectrum of clinical presentations of antibody-mediated rejection (AMR). AMR can only be diagnosed when the four criteria are met. The presence of donor-specific antibodies (DSAs) poses a risk of graft injury. By itself, the deposition of C4d is not a criterion for AMR. If DSAs and C4d are identified simultaneously to vascular histological injury, there is a high risk of graft dysfunction
AMR can cause two different forms of graft dysfunction: classic arterial hyperacute AMR and vascular AMR. Classic arterial hyperacute AMR is characterized by necrotizing arteritis, with mural fibrinoid necrosis and inflammation in artery walls. Endothelial cells are damaged and luminal thrombosis is common, resulting in organ infarction. Hyperacute rejection occurs minutes after transplantation and is caused by preexisting Abs against the donor . These Abs activate the classical pathway of the complement system and initiate the blood clotting cascade. Vessels become obstructed and neutrophils are rapidly recruited to the allograft. Hyperacute AMR differs from Ab-mediated vascular rejection in that it does not feature an inflammatory or fibrinoid component in vessel walls as its outset. Histological evidence of tissue injury in the context of vascular AMR usually includes the presence of neutrophils and macrophages in capillaries, endothelial damage such as swelling and denudation in cardiac allografts, fibrinoid necrosis, thrombi, and acute tubular injury in kidney allografts. Vascular AMR is generally characterized by abundant deposition of the complement activation product C4d in peritubular capillaries (Fig. 12.2) [44, 23, 24, 6, 26, 54].
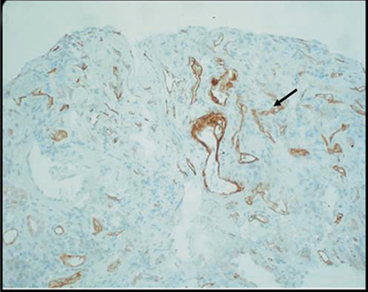
Fig. 12.2
Renal allograft biopsy from a child with chronic allograft nephropathy showing C4d deposition (brown) in peritubular capillaries consistent with humoral-mediated rejection (reprinted with permission, Fletcher JT)
A significant number of episodes of AMR occur in sensitized patients, in whom donor-specific antibodies (DSAs) are already present in the bloodstream. DSAs may form after implantation of the allograft, or may be present before transplantation. They may arise in patients who have been pregnant or have undergone transfusions or prior transplants. Mismatched blood type (ABO-incompatible) transplants may be subject to DSA-mediated AMR and display features of the phenomenon termed accommodation [56, 49].
Hyperacute rejection occurred more frequently before the introduction of crossmatch (CXM) testing. CXM testing is used to detect the presence of Abs against the lymphocytes of an individual donor. A positive T-cell CXM test is usually considered an absolute contraindication to kidney transplantation because of the high risk of hyperacute rejection. Although CXM testing initially presented low sensitivity, the addition of antihuman globulin (AHG), flow cytometric crossmatch (FXM) testing and solid-phase beads have since improved its sensitivity: it is now capable of detecting anti-class-I and class-II Abs. The panel-reactive Ab (PRA) assay is a screening test used to measure breadth of sensitization as determined by the presence of anti-HLA Abs [27]. Since the enhancement of the PRA assay by the addition of AHG and flow cytometry, it is now able to detect very low levels of Abs and noncytotoxic Abs. The PRA tests the recipient’s serum for its ability to lyse a panel of T lymphocytes that is a surrogate of potential donors. Despite continuous improvements in the technique, the panel does not reflect all potential donors and provides limited information about the specificities of the Abs .
Recipients with a positive FXM test after a negative CXM test are at very low risk of hyperacute rejection. However, they are at an increased risk of AMR, CMR, or both. The introduction of solid-phase assays permits identification of the presence of anti-HLA Abs against a wide range of HLA types and determination of their HLA specificity. The detection of DSAs in the CXM or solid-phase assays is not considered an absolute contraindication to kidney transplantation, but it represents an immunological risk of Ab-mediated injury. Whereas a positive CXM test and high DSA levels pose a notable risk of hyperacute rejection, a positive CXM test coincident with low levels of DSAs may increase the risk of AMR [25].
The presence of preformed Abs at the time of kidney transplantation was found to be associated with chronic allograft failure (CAF), which is the second leading cause of kidney allograft loss after death [91]. Up to 60 % of patients diagnosed with CAF show evidence of Ab-mediated injury. Although anti-HLA Abs are usually detectable several weeks before graft dysfunction, they are not the sole cause of AMR [91]. A study that followed 70 kidney transplants demonstrated that AMR occurs across a wide spectrum of baseline DSA levels, even after a negative T-cell AHG CXM test. The risk of AMR increased with increasing baseline levels of DSA, but its occurrence remained unpredictable. Interestingly, prior transplantation did not increase the incidence or severity of AMR compared with other methods of sensitization. The authors concluded that anti-class-II DSAs, alone or in combination with anti-class-I DSAs, played an important role in AMR and could be the sole cause of AMR in as many as 16 % of cases [11] .
The detection of C4d deposits is considered a vital tool for the diagnosis of AMR. Before identification of the C4d component, many Ab-mediated rejection episodes went undiagnosed. Attempts to diagnose humoral rejection were unsuccessful, probably because of the rapid endocytosis of Abs from the endothelial surface [23]. Current immunopathological evidence of AMR includes positive staining for complement products such as C1q, C3d, or C4d, which may also be accompanied by Igs G, M, and A [80, 23]). C4d in transplanted kidneys is characteristically located in the peritubular capillaries, [95] and should be considered the ‘fingerprint’ of an Ab response [7]. However, a variety of situations can accompany the localization of C4d in peritubular capillaries. Whereas up to 90 % of C4d + cases exhibit anti-HLA Abs, in 10 % of C4d + recipients, the search for DSA yields no results (Fig. 12.3) [56]. This may be due to the technical limitations of assays or the presence of unusual Abs. Conversely, there is emerging evidence of episodes of AMR that are C4d − in the kidney and in other allografts [72]. Indeed, a recent study showed that a high percentage of recipients presented circulating DSAs that were not associated with C4d deposits [85, 56]). An explanation for this phenomenon might be that the pattern of C4d deposition is dynamic and can disappear after a few days. Deposition of C4d can also persist, despite anti-rejection therapy [56].
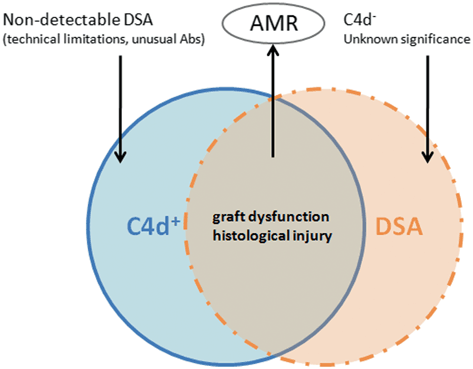
Fig. 12.3
Humoral rejection needs be diagnosed in the presence of antibodies and C4d. Other possible presentations may occur. AMR antibody-mediated rejection; DSAs donor specific antibodies
C4d is not an exclusive marker of AMR. It has also been found in protocol biopsies of stable grafts and is not even a molecule limited to allografted organs; it has been found in biopsies of normal kidneys, located at every glomerular mesangium, and at the vascular pole of the arterioles. The accumulation of C4d in glomerular capillaries has also been implicated in kidney diseases caused by the formation of immune complexes [56].
Liver allografts are less susceptible to AMR than other solid organ allografts. This has been attributed to phagocytosis by Kupffer cells of immune complexes generated after Ab binding and the absence of a basal membrane in the sinusoidal hepatic microvasculature [22]. Following liver transplants, anti-HLA Abs do not always cause clinically relevant allograft damage, whereas isoagglutinins usually cause more damage. Hyperacute rejection is followed by liver dysfunction over a period of hours to days. This can be recognized by swelling, a dusky appearance of the liver, cessation of bile flow, difficulty in achieving hemostasis, and an exaggerated need for platelets and blood replacement. Arterial thrombosis is evident on post-transplant angiograms. Areas of necrosis are visible in gross examinations of failed liver grafts. Histological examination reveals findings common to other organs, such as vessel congestion, thrombi, and necrotizing arteritis. For “vascular” humoral rejection caused by preformed anti-HLA Abs, the changes are usually less florid and necrotizing arteritis is rare. Detection of immune reactants relies on the localization of C4d deposits. However, C4d is also evident during CMR [22, 20].
In cardiac transplantation, AMR has only recently been recognized as a real and distinct clinicopathological entity [42]. Hemodynamic dysfunction has been attributed to AMR more frequently in women than in men. The presence of DSAs is strongly correlated with AMR and decreased allograft and patient survival [62]. Macroscopically, the heart is swollen and discolored, showing areas of focal necrosis. Histological examinations frequently reveal intravascular thrombi and endothelial swellings. There are abundant accumulations of macrophages in the capillaries. Mixed AMR and CMR rejection is evident in 15 % of biopsies of hemodynamically abnormal cardiac allografts [47]. Similarly to the kidney, C4d deposition is a footprint of AMR in the heart. It has been associated with a poor prognosis but deposition of C4d alone should not be equated with AMR [42]. In lung transplants, histopathological criteria have not yet been established for the diagnosis of AMR .
Cellular and Molecular Basis of Antibody-Mediated Rejection
Only B-lymphocytes are able to generate humoral immunity . They originate in bone marrow and are divided into two subsets; whereas the B1 lineage contributes to innate immunity and usually produces Abs against microbes, the B2 lineage forms part of the adaptive immune system that resides in the bloodstream and peripheral lymphoid tissues. Each B cell produces only one specific type of Ab, placing a transmembrane molecule on its surface to act as receptor for the antigen (Ag) specific to that Ab. The B cell places approximately 105 identical Ab molecules on its surface [21]. If an Ag binds one of these molecules while the B cell resides in the secondary lymphoid tissues, it transforms into a plasma cell (PC). PCs are able to secrete a soluble version of the specific Ab with the same specificity as the membrane-bound Ab characteristic of the B-lymphocyte .
Only B lymphocytes that bind their specific Ag proliferate in successive waves; this phenomenon is known as clonal selection. Other cells, such as dendritic and T-helper follicular cells, aid the formation of a germinal center of activated B cells. It takes several days before Abs are detectable in the serum following primary contact with an Ag. This type of response to antigens is termed the acquired immune response. The maturation of B cells changes the expression of cell-surface markers. For example, the expression of CD20 and CD19 is high on unstimulated B cells, but low-to-absent on PCs. Additional markers of the later stages of B-cell development include B cell activating factor from the tumor necrosis factor family (BAFF), A proliferation inducing ligand (APRIL) and Blimp-1 [76]. Identification of B cells requires more than one marker because of the variability of B-cell phenotypes; for example, not all memory B cells express CD20. The final step in the development of persistent Ab production involves the migration of PCs back to the bone marrow. Most PCs express CD138 and CD38; these markers are not expressed on unstimulated B cells. PCs that produce Abs against specific HLA are rare, with an average frequency of a single HLA-specific PC per 2 × 106 bone marrow cells [61].
The complement system comprises 20 plasma proteins that are activated in a cascade-like manner (Fig. 12.4). There are different ways of activating this cascade, including the perception of microbial polysaccharides, Ab binding, and the mannose–lecithin pathway. Complement activation results from the deposition of molecules that can produce direct lysis, facilitate opsonization, and enhance phagocyte function; byproducts of the complement system act as chemotactic factors and activate mast cells . The complement components are named using the letter ‘C’ followed by a number related to the chronology of its discovery.
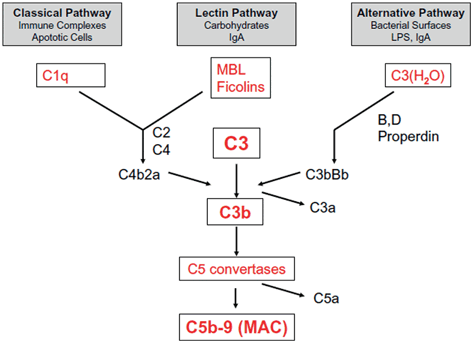
Fig. 12.4
Schematic representation of the complement system. Ig immunoglobulin; LPS lipopolysaccharide; MAC membrane-attack complex; MBL mannose-binding lectin. (Reprinted with permission, Berger ST)
AMR damage is caused by classical complement activation when donor-reactive HLA Abs bind to allograft endothelial cells. The Abs bind C1qrs, which in turn binds C4; C4 cleaves to C4b, then recruits C2 and C3 to form C4b2a, a convertase of C3. Then, it forms C4b2a3b, which cleaves C5 into C5b, leading to the formation of the annular membrane-attack complex (MAC) C5b-9. The MAC is a transmembrane channel that is fully permeable to electrolytes and water. Owing to the high internal colloid osmotic pressure of cells, there is a net influx of Na + and water into cells that contain the MAC; this causes cell lysis. The C5-9 MAC also mediates neutrophil influx and synthesis of pro-inflammatory cytokines , causing cell injury, apoptosis, and necrosis. The C5a receptor on endothelial cells seems to function in the production of adhesion molecules and may regulate apoptosis [25].
In contrast, C4d is an inert degradation product of C4 that binds the cell surface. Complement component C4d was linked to the presence of DSAs and AMR in the 1990s and has since been considered a footprint of the presence of humoral rejection. C4d is a marker of complement cascade activation that is evident in the endothelium of peritubular capillaries and basal membrane of kidneys (Fig. 12.2) and in the portal capillaries of livers undergoing humoral rejection. Histological tests are best performed on frozen section specimens [91, 4]. Using immunohistochemical techniques, donor-specific IgG and IgM are undetectable on the vascular endothelium of renal allografts, even in the context of AMR [25].
Antibody-Mediated Rejection and Accommodation
Clinical and experimental research has demonstrated that allografts can develop resistance to anti-graft Abs . This is best established in ABO-incompatible kidney allografts and is usually referred to as accommodation [19, 49]. However, the mechanisms involved are not well understood.
It has been suggested that accommodation might result from a change in the expression of Abs from a complement-fixing form to a non-fixing form, or from the lack of subsets of T-helper cells [52, 81]. However, no conclusive data explain accommodation as a result of the modification of allograft antigens. Accommodated allografts seemed to change their phenotypic profile compared with ABO-compatible grafts. The evidence was stronger for ABO-incompatible grafts than for renal allograft recipients with positive-CXM tests [59]. In contrast, others have suggested a resistance to complement that might reflect modification of the complement cascade such that its effector functions cause little or no injury to graft endothelium [49]. This may be the result of complement regulation, resistance to lysis, and improved metabolism of complement complexes.
Accommodation may be more frequent than expected in functioning allografts; there is a possibility that Abs bind to the allograft that will subsequently be removed. This is consistent with the clinical finding of increased serum Abs after failed allograft removal [49].
There is a paucity of reported strategies to facilitate accommodation. Using an experimental model, Wang and colleagues reported that administration of anti-C5 Ab in presensitized allograft recipients, in conjunction with immunosuppression, prevented rejection and permitted accommodation. However, double-transplant experiments demonstrated that immunological alterations in both the graft and recipient were required for successful graft accommodation [86].
Prevention and Treatment of Antibody-Mediated Rejection
A complete patient sensitization history, which includes PRA results, CXM test results, prior transfusions, pregnancies, and previous transplants is required to assess the risk of AMR for renal, cardiac, and lung transplants . An individual cannot be reliably defined as unsensitized without a complete sensitization history [80]. Sensitive techniques should be used to determine the presence or absence of DSAs, and the specificity of DSAs should be determined before transplantation. The presence or absence of auto-Ab must be established to facilitate the interpretation of pretransplant CXM test results. In addition, both T- and B-cell CXM tests should be performed before transplantation (unless clinically contraindicated, i.e., after a long period of cold ischemia—and only then if a complete and reliable sensitization history is negative) [80]. The risk of AMR and early graft loss in patients showing a positive complement-dependent cytotoxicity (CDC) on CXM testing is high. Transplantation should be avoided unless a desensitization protocol can be used. A positive CXM test using the flow cytometry technique poses medium risk and requires high doses of immunosuppression. Recipients with negative flow cytometry results or CDC on CXM testing are at low risk of AMR.
AMR can be managed in different ways, depending on the treatment target, and some therapies can tackle several of these targets. Plasmapheresis (PP) and immunoadsorption (IA) can control B-cell responses by removing the Abs against donor antigens, whereas administration of intravenous Ig (IVIG) dilutes alloantibodies, which mediate the effector arm of AMR. Alternative approaches are to neutralize B-cell division with mycophenolate mofetil (MMF) or inhibit B-cell surface molecules with rituximab. On the other hand, the use of MMF and steroids to inhibit T-cell division, CNI to prevent the signaling of interleukin (IL)-2 to T cells, or anti-thymocyte globulin (ATG) to ablate T cells can restrain T cells from activating B cells. Additional drugs, such as bortezomib, act against PCs, inhibiting what is considered the basis of AMR. There is also an arsenal of drugs that prevent the fixation of complement, including Abs such as eculizumab, which decreases the spread of the complement cascade. Finally, several of these steps can be circumvented by a nonspecific treatment, such as splenectomy, which limits the development of B cells [45, 78, 5].
ATG is a polyclonal Ab preparation generated from the immunization of rabbits with human thymus. ATG has different effects on cells involved in rejection, such as anti-T-cell effects and inhibition of the interaction between CD4 + helper T cells and B cells; these reduce rates of B-cell activation. However, ATG may also have direct cytotoxic effects on B cells, both by modulating the production of allo-Abs and by causing the apoptosis of B cells. These effects are due to different specificities against T cells (class-I and class-II HLA molecules, CD3, CD4, CD8, costimulatory pathways and cell adhesion molecules), B cells (CD20) and PCs (CD38 and CD138) [8, 9, 94]. This therapeutic option is included in different treatment algorithms, particularly when there is histological evidence of combined cellular rejection and AMR [74, 65, 71, 87, 3, 40].
IVIG is derived from the plasma of thousands of healthy blood donors and is primarily composed of IgG (90 %), a few dimers, Ag-binding fragments and traces of IgA and IgM [48]. In the 1990s, IVIG was discovered to inhibit the cytotoxic effects of allo-Abs [30, 83]. Nowadays, IVIG is used in desensitization protocols and for the treatment of AMR. IVIGs have different specificities against class-I and class-II HLA molecules, CD 40 costimulatory molecule, IL-1, IL-4, IL-6, and cytokines such as tumor necrosis factor-α, T cells and interferon-γ receptor among others [28]. IVIG has several immunomodulatory effects on activation of the complement system, including binding and blockading Fc receptors, regulating the mechanism of immune responses by diminishing allograft rejection by dendritic cells, inhibiting T-cell activation by dendritic cells, B-cell apoptosis, and downregulating the B-cell receptor [2, 82, 93, 41, 73, 89, 67, 37, 88]. Common adverse effects include headache, fever, myalgia, arthralgia, chills, hypotension, and hypertension. Slowing the infusion rate and using iso-osmolar preparations can reduce these adverse effects. Serious adverse effects include aseptic meningitis, acute renal failure related to high osmotic load, anaphylaxis associated with IgA sensitization in recipients with IgA deficiency, and thrombotic events related to rapid infusion rates [41, 18, 63, 36].)
PP acts to remove allo-Abs from the circulation and is the fastest, most effective therapeutic option for reducing the levels of DSAs. It is used in both desensitization protocols and in AMR treatment [17, 65, 60, 53]. Some of the currently available variants of PP are plasma exchange, double filtration and IA PP. These treatments are usually applied in combination with either IVIG, rituximab, MMF or CNIs, or bortezomib. Adverse effects related to PP, which occur in 5–12 % of all patients, are considered to be mild or moderate. They include volume concentration, bleeding diathesis, allergic reactions, and blood-borne pathogen transmission [92, 77, 58].
Rituximab is a chimeric monoclonal Ab against anti-CD20, an Ag expressed in the early stages of the B-cell cycle, and is absent on mature PCs. CD20 regulates the early steps involved in initiation of the cell cycle and cell differentiation. Rituximab comprises a human IgG1 heavy chain and κ-light chain constant region fused with mouse variable regions. The mechanisms of rituximab-induced depletion are Ab-dependent T-cell-mediated cytotoxicity, complement-mediated cell killing, and induction of apoptotic cell death via CD20 [90, 31, 84, 13, 1, 16]. Rituximab has been successfully used as induction therapy for sensitized patients; however, few protocols have used it to treat AMR [68, 87]. This agent produces a marked depletion of circulating B cells and a less marked reduction of B cells present in the spleen and lymph nodes [3, 64]. Rituximab has no effect on PCs, which are the source of DSAs; accordingly, its effect on the production of allo-Abs has no relationship with the depletion of Ab-producing cells. The therapeutic benefit of rituximab may be related to depletion of B memory cells and the modification of cellular immunity, rather than a reduction in the abundances of Abs [31, 12, 75, 66, 32]. A single dose in renal transplant patients can result in prolonged B-cell depletion, with populations that are suppressed for 1–2 years [29].
Bortezomib is a proteasome inhibitor that causes apoptosis of PCs, reducing allo-Ab production in sensitized patients [46]. Bortezomib binds to the 26S proteasome, which is part of an enzyme complex that is the primary proteolytic mechanism of the eukaryotic cell [10
 Bayesian Classifier and Molecular Marker Platforms for Immune Monitoring
Bayesian Classifier and Molecular Marker Platforms for Immune Monitoring
 Immunological Similarities and Differences Between Extremity and Face Transplants
Immunological Similarities and Differences Between Extremity and Face Transplants
 Mesenchymal and Adipose Stem Cell Strategies for Peripheral Nerve Regeneration
Mesenchymal and Adipose Stem Cell Strategies for Peripheral Nerve Regeneration
 Advances in Diagnosing Skin Rejection and Immune Monitoring
Advances in Diagnosing Skin Rejection and Immune Monitoring
 Mesenchymal Stem Cells as Immune Modulators in VCA
Mesenchymal Stem Cells as Immune Modulators in VCA
 Experimental Models and Clinical Tools to Assess Nerve Regeneration and Functional Outcomes
Experimental Models and Clinical Tools to Assess Nerve Regeneration and Functional Outcomes




Related posts:
Bayesian Classifier and Molecular Marker Platforms for Immune Monitoring
Immunological Similarities and Differences Between Extremity and Face Transplants
Mesenchymal and Adipose Stem Cell Strategies for Peripheral Nerve Regeneration
Advances in Diagnosing Skin Rejection and Immune Monitoring
Mesenchymal Stem Cells as Immune Modulators in VCA
Experimental Models and Clinical Tools to Assess Nerve Regeneration and Functional Outcomes
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
