Adamantiades–Behçet Disease: Introduction
|
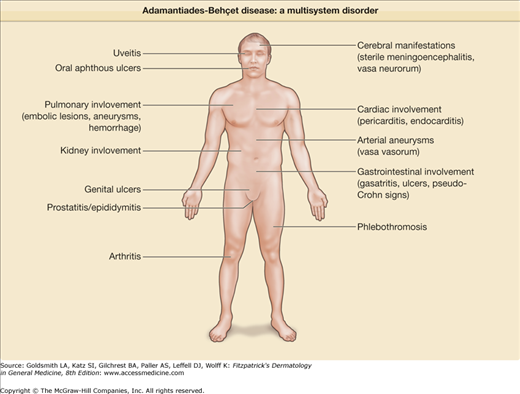
Adamantiades–Behçet disease is a multisystem inflammatory disease of unknown etiology, classified as systemic vasculitis involving all types and sizes of blood vessels and characterized clinically by recurrent oral aphthous and genital ulcers, skin lesions, and iridocyclitis/posterior uveitis, occasionally accompanied by arthritis and vascular, gastrointestinal, neurologic, or other manifestations1,2 (Fig. 166-1).
Historical Aspects
Hippocrates of Kos (460–377 bc) used the designation “στoματα αϕθωδϵα, ϵλκωδϵα” (oral aphthous ulcers) in a probable first description of a patient with the disease (Epidemion Book III, Case 7). The disease is named after Benediktos Adamantiades, a Greek ophthalmologist and Hulûsi Behçet, a Turkish dermatologist, who, in 1931 and 1937, respectively, described patients with the characteristic clinical complex insisting for a single clinical entity.3 The first international multidisciplinary conference was organized by two dermatologists, Drs. M. Monacelli and P. Nazarro, 1964 in Rome, Italy.
Epidemiology
Adamantiades–Behçet disease presents a worldwide occurrence with varying prevalence, being endemic in the Eastern and Central Asian and the Eastern Mediterranean countries (along the so-called Silk Road) and rare in Northern European countries, Central and Southern Africa, the Americas, and Australia.4 A prevalence of 80 to 420 patients per 100,000 inhabitants has been reported in Turkey,5 7 to 30 patients per 100,000 inhabitants in the rest of the Asian continent (Japan, 14–31:100,000; Korea, 7.3:100,000; Northern China, 14:100,000; Saudi Arabia, 20:100,000; Iran, 17:100,000) and 1.5–7.5:100,000 in Southern Europe. In Northern Europe (0.27–1.18:100,000) and the United States (0.12–0.33:100,000), the disease is rare.4 In countries with several ethnic populations, including of Turkmen and Mongol descents, the latter are mainly affected. The increasing prevalence of the disease is due to its chronic character. Its annual incidence is low; 0.75 to 1.0 new cases per 100,000 inhabitants were assessed in Japan (1990) and Germany (2005).6 Adamantiades–Behçet disease most often affects patients in their 20s and 30s; however, early and late onsets (first year of life to 72 years) have been reported. Juvenile disease rates are 2 to 21% in different ethnic groups; its prevalence was estimated to be 0.17:100,000 in France. In contrast to old Japanese and Turkish reports of male predominance, the male-to-female ratio drastically decreased in the last 20 years. Currently, both genders are equally affected; a male predominance is still observed in Arab populations, whereas female predominance is evident in Korea, China, some Northern European countries, and the United States.
Etiology and Pathogenesis
The etiology of the disease remains unknown, although genetic factors, infectious agents, environmental pollution, immunologic mechanisms, and endothelial and clotting factors have been implicated and studied intensively.7,8 The endemic occurrence along the historical Silk Road, the major involvement of certain ethnic groups (mostly of Turkmen and Mongol descent), and associated immunogenetic data support the hypothesis that the disease followed the migration of these old nomadic tribes. On the other hand, the wide variation of the disease prevalence in the same ethnic group in association with different geographic areas of residence indicates an additional environmental triggering factor. Therefore, transfer of genetic material and/or of an unknown exogenous agent may have been responsible for the expansion of the disease.
There is no specific mode of Mendelian transmission in Adamantiades–Behçet disease.7,8 Familial occurrence with regional differences has been reported, being more frequent in Korea (15%) than in Japan or China (2–3%) and in Arab countries, Israel, and Turkey (2–18%) than in Europe (0–5%). An earlier disease onset in children compared with their parents and a higher frequency of familial cases in juveniles than in adults has been observed.
A significant association exists between the disease and human leukocyte antigen-B51 (HLA-B51) in Japan, the Middle East, and the Mediterranean countries; however, this relationship is not as strong in Western countries.9 The allele also seems to be associated with a more severe prognosis.10 Its exact role in the disease mechanism is still unknown; however, it may be involved in the disease development through specific antigen presentation, molecular mimicry with microbial antigens, or participation in linkage disequilibrium with a presently unknown susceptibility gene.11,12 Among the 24 currently described alleles, HLA-B5101 and -B5108 have most frequently been associated with Adamantiades–Behçet disease.13 Shared amino acid residues (defining the Bw4 epitope) are crucial for antigen binding and natural killer cell interactions,14 and Bw4 was also reported to contribute to the severity of the disease.15 Genes possibly associated with the disease have been localized on chromosome 6 in the region between the tumor necrosis factor gene and HLA-B or HLA-C genes, including the major histocompatibility complex class I chain A gene (A6 allele) and genes for heat shock proteins8,11,13,16 (eFig. 166-1.1). In addition, a novel susceptibility locus mapped to 6p22–23 was detected.13 Lately, associations on chromosomes 1p31.3 [Interleukin (IL) 23R-IL12RB2] and 1q32.1 (IL10) were found by genome-wide association studies.14,17 A haplotype association of IL-8 gene with Adamantiades–Behçet disease was also detected.18 Polymorphisms in gene encoding for host effector molecules may contribute to the disease susceptibility to and/or severity of the disease, such as in IL-23R reported in a Chinese Han population.19
eFigure 166-1.1
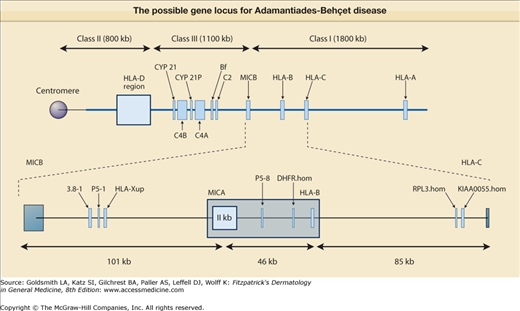
The possible gene locus for Adamantiades–Behçet disease (Online Mendelian Inheritance in Man #109650) is located in the area of the major histocompatibility system (i.e., on chromosome 6p21.3). MICA = major histocompatibility complex class I chain A; MICB = major histocompatibility complex class I chain B. (From Zouboulis CC, May T: Pathogenesis of Adamantiades-Behçet’s disease. Med Microbiol Immunol 192:149, 2003, with kind permission of Springer Science and Business Media.)
Adamantiades–Behçet disease is not considered contagious as no horizontal transmission has ever been reported. However, viral and bacterial infections have been implicated in initiating immunopathologic pathways, leading to the onset of the disease.7,8
Early theories of the pathogenesis of Adamantiades–Behçet disease proposed a viral or other infectious etiology.3 Partial transcription of herpes simplex virus type 1 (HSV-1) DNA in patients’ peripheral blood lymphocytes was reported.7 HSV-1 DNA was detected in patients’ saliva and oral and genital ulcers, and HSV-1 antibodies were found in patients’ serum.
The disease activity has been known to correlate with bacterial infection, particularly Streptococci.7 Streptococcus sanguinis (S. sanguinis) dominates the flora of the oral mucosa in patients with the disease and appears to be the most relevant Streptococcus strain as a provoking factor for initiation of the disease.20 Streptococcus antigens and antistreptococcal antibodies are frequently found in the oral mucosa and serum of patients. The involvement of immunoglobulin A protease-producing S. sanguinis is proposed as an explanation for a chronic infection leading to initiation of Adamantiades–Behçet disease. High titers of the immunogenic S. sanguinis antigen KTH-1 have been detected in patients. In addition, exposure of the patients to Streptococcus antigens may be a major provoking factor for disease activity. The lipoprotein of Mycoplasma fermentans MALP-404 was found in the serum of patients with Adamantiades–Behçet disease but not in healthy controls.21 Interestingly, MALP-404 contains a peptide motif, which can be presented by HLA-B51. A possible role for bacterial stimulation of monocytes via Toll-like receptor-2 producing neutrophil-stimulating proinflammatory factors in Adamantiades–Behçet disease was currently detected.22
Immunologic mechanisms are considered to play a major role in the pathogenesis of Adamantiades–Behçet disease.7,8,13,16 The disease has currently been classified among the autoinflammatory disorders,23 which are caused by primary dysfunction of the innate immune system.
The major microscopic finding at most sites of active disease is an immune-mediated occlusive vasculitis. The pathergy reaction (see Section “Clinical Findings”) is induced by the rapid accumulation of neutrophils (hyperchemotaxis) and later by T lymphocytes and monocytes/macrophages at the needle prick sites. T cells in the peripheral blood and in the involved tissues are increased, and a predominant T helper 1 immune response induced by IL-12 has been demonstrated.24 Patients’ lymphocytes also express CD29 molecules and bind to endothelial cells in active disease. In addition to defective T-cell immunity, B-cell activation is impaired. Circulating immune complexes, together with enhanced neutrophil migration, may be involved; diversity of T cells indicates that specific T-cell responses to several antigens may lead to the variety of symptoms.25 Tropomyosins and the 160-kDa polypeptide kinectin have been detected as autoantigens in Adamantiades–Behçet disease.26,27
Increased levels of heat shock protein (HSP)-specific antibodies in serum have been found in Adamantiades–Behçet disease.28,29 T cells respond to 60-kDa HSP, and four different peptide determinants within 60-kDa HSP identified by T-cell epitope mapping have been suggested to be involved in the pathogenesis of the disease.
Various proinflammatory cytokines, such as IL-1, -8, -12, -17, -23, and tumor necrosis factor-α, are elevated in the sera of patients with Adamantiades–Behçet disease.30–34 In particular, IL-8 seems to play an important role, can also be released by endothelial cells, has a potent effect on the inflammatory response, and is a sensitive marker of disease activity.30–32 Cytokine release may be dependent on the involved organ.30,31,33
The endothelium seems to be the primary target; however, it may just be subject to the bizarre behavior of the immune system.35 An immunoglobulin M-type, 47-kDa cell surface HSP against endothelial α-enolase was identified in the serum of patients with Adamantiades–Behçet disease.36 Plasma endothelin-1 concentrations were found significantly increased, perhaps indicating vasoconstriction and being the direct result of elevated synthesis by injured vascular endothelial cells. Thrombomodulin, a cell surface glycoprotein of vascular endothelium, which is also increased in the plasma of patients with active disease, potentially damages the endothelial cells.
Clinical Findings
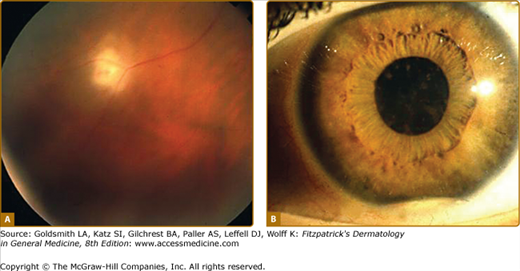
Adamantiades–Behçet disease is a chronic, recurrent, multisystem, and, occasionally, life-threatening disorder.1,6 Recurrent oral aphthous ulcers, recurrent genital ulcers, skin manifestations, ocular lesions, and arthritis/arthropathy are the most frequent clinical manifestations. Vascular, gastrointestinal, neurologic, psychiatric, pulmonary, renal, and cardiac manifestations; epididymitis; and other findings can also occur. The clinical picture usually develops within a few months after the presenting sign; both an acute multisystem presentation and long-term development of the disease over years are possible.
Diagnosis of Adamantiades–Behçet disease is based on clinical signs, as pathognomonic laboratory test or histologic characteristics are absent. There are several sets of diagnostic criteria, the most popular of them being the criteria of the International Study Group37 and those of the Behçet Disease Research Committee of Japan.38 However, there have been several problems with these criteria, including their performance in selectivity and specificity, so that both of them have currently been revised2,39,40 (Tables 166-1, 166-2).
|
Symptom | Points |
|---|---|
Ocular lesions (recurrent) | 2 |
Oral aphthosis (recurrent) | 2 |
Genital aphthosis (recurrent) | 2 |
Skin lesions (recurrent) | 1 |
Central nervous system | 1 |
Vascular manifestations | 1 |
Positive pathergy testb | 1 |
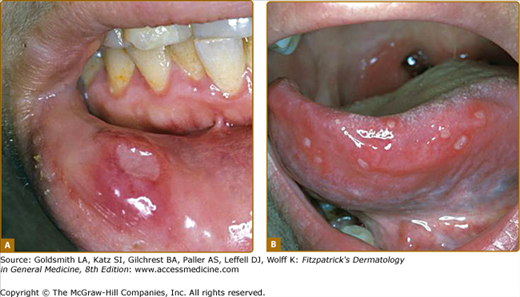
Recurrent oral aphthous and genital ulcers are the most frequently observed mucosal manifestations. Oral aphthous ulcers are the presenting sign in more than 80% of the cases.1,6,16 Although recurrent aphthous stomatitis is a common disorder, only a few patients progress to Adamantiades–Behçet disease, and it is not possible to determine in whom or when the transition may occur.41 Typically, lesions are multiple, painful, 1–3 cm in diameter, and sharply margined with a fibrin-coated base and surrounding erythema (Fig. 166-2). Oral aphthous ulcers usually heal without scarring (92%). Genital ulcers may not recur as often and usually heal with a characteristic scar (64%–88%; Fig. 166-3). Spontaneous healing of aphthae occurs within 4 days to 1 month; genital ulcers may persist longer. Large oral ulcerations can also be associated with problems such as pharyngeal involvement, dysphagia, and dyspnea or fistulae involving the pharynx, larynx, trachea, or esophagus. Genital ulcers can occur on the penis, scrotum, vagina, labia, and urethra, and also in the anal, perineal, and inguinal regions.
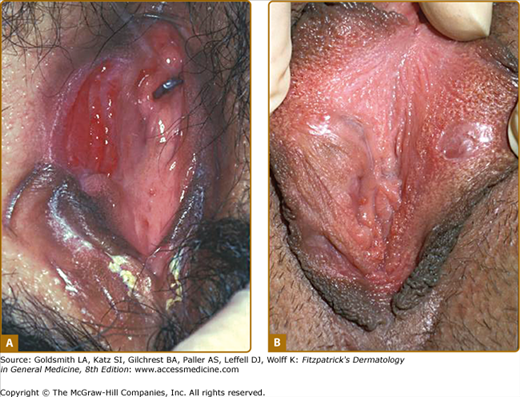
Skin lesions that should be accepted as diagnostically relevant in Adamantiades–Behçet disease should be confined to pustular vasculitic lesions (including pathergy lesions), erythema nodosum-like lesions, Sweet-like lesions, pyoderma gangrenosum-like lesions, and palpable purpuric lesions of necrotizing venulitis (see Fig. 166-5). All of these lesions are characterized in their early stages by a neutrophilic vascular reaction. Acneiform lesions or follicle-based pustules should not be considered relevant.42
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


