Introduction
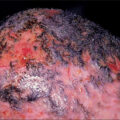
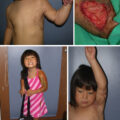
Despite ongoing research and an increased understanding of the underlying factors, hypertrophic scar (HSc) continues to plague burn survivors and challenge burn surgeons. This chapter is an incremental evolution from previous editions of this textbook, reflecting the incremental changes in our understanding of HSc, and we are indebted to the previous authors, including Dr. Alexis Desmoulière, for their insights. Clinically, physicians distinguish HSc as scar that is elevated, erythematous, and inelastic ( Fig. 44.1 ) as compared with normotrophic mature scar (NSc), which is flat, soft, and pliable. Meanwhile, patients experience HSc as uncomfortable, sometimes painful, pruritic, inelastic, and prominent scar that has poor cosmesis and often impair function through contractures and decreased range of motion. The crucial differences between normal skin and mature scar vs HSc fall into three main categories: altered extracellular matrix (ECM) content and structure, abnormal keratinocytes and fibroblast phenotype, and an imbalance of profibrotic cytokines and signaling factors. Just as scars in general are dynamic and then mature over 1 to 2 years, so too HSc also undergoes dynamic changes to some degree over time, which leads to variability in descriptions of HSc and the resulting research. Unfortunately, without intervention, HSc does not mature to the degree that NSc does, and ultimately causes long-term morbidity for patients.
HSc is one of many fibroproliferative disorders and shares many features with scleroderma, pulmonary fibrosis, and renal fibrosis. Thus, treatments for these other disorders provide new avenues of investigation for improving HSc, and increased understanding of HSc pathophysiology often moves the field forward for all fibroproliferative disorders.
Although we will discuss the various molecular and cellular factors of HSc one at a time, this is an artificial approach, and the reality is that they combine and interact in complex networks that ultimately result in HSc. Unfortunately, this is one of the reasons that no silver bullet or magical cure for HSc has emerged despite intensive research into fibrosis and scarring in humans. To maintain relevance, this chapter focuses specifically on well-established HSc pathways, but it is neither exhaustive nor encyclopedic. It is our continued hope that an ever-increasing understanding of the pathophysiology of abnormal wound healing will lead to effective and predictable treatments not only for burn patients suffering from HSc but also for the many other patients with fibrotic diseases as well.
Extracellular matrix content and structure
ECM is produced and structured by fibroblasts during growth, development, wound healing and scar maturation. ECM in HSc is significantly different from that in NSc and normal skin, primarily in collagen bundle composition and arrangement, and in the relative proportions of multiple proteoglycans and glycoproteins. Not only do fibroblasts produce ECM, but they also respond to their ECM microenvironment, thus HSc, the ECM alterations also contribute to abnormal fibroblast behavior, with the potential to create a profibrotic feedback loop.
Collagen and extracellular matrix morphology
Human ECM is primarily composed of various types of collagen, arranged in fibrils that provide both a cellular scaffold and mechanical strength to the tissue. Because of the increased thickness of HSc, there is an increase in the quantity of collagen per unit surface area, but the overall density of collagen is lower than that in normal skin because of a relatively greater increase in proteoglycan (GAG) and glycoprotein content. In normal skin, type I collagen is predominant (80%), with smaller amounts of type III (10%–15%) and type V (minimal) collagen, which are arranged in thick, regular collagen fibril bundles that run parallel to the skin surface. In HSc, there is significantly more type III (33%) and type V (10%) collagen, with thin and disorganized collagen fibrils. In healing wounds that result in NSc, type III collagen appears early then gradually disappears as the scar remodels and matures, but in HSc it persists in high levels suggesting biologic immaturity.
As mentioned, the morphologic structure of HSc is drastically altered from that of normal skin. Although normal skin has thick collagen bundles arranged in a basket-weave pattern, HSc has whorls and nodules of fine collagen. This altered morphology is virtually pathognomonic of HSc and is the result of alterations in collagen, proteoglycan composition, and their relative ratios. This altered morphology is not only visually obvious but can also be mathematically described using the collagen orientation index. Not only does this abnormal morphology reflect the underlying alterations in collagen composition but also results from significant differences in proteoglycans and glycoproteins, which fill the interfibrillar space.
Proteoglycans and glycoproteins
Proteoglycans interact with collagen fibrils and contribute to many of the physical properties of skin, including turgor, resilience, and resistance to compression. Proteoglycans also modulate multiple growth factors and cytokines, and they influence collagen fibril spacing and compaction. The glycoproteins such as fibronectin, are involved in cell matrix adhesion, and together with proteoglycans they are major components of skin, influencing both function and structure.
The proteoglycans consist of a protein core and glycosaminoglycan side chains, which are ionized and hydrophilic, leading to tissue water retention. The increased proteoglycan content in HSc causes hyperhydration, which increases tissue turgor. In HSc, as compared with normal skin, the profrotic proteoglycans versican and biglycan are significantly upregulated, whereas an antifibrotic proteoglycan decorin is downregulated. Decorin is a small leucine-rich proteoglycan present in large amounts in normal skin and NSc, but reduced by 75% in HSc. Decorin binds to collagen fibrils and controls their diameter, morphology, and spacing. Decreased decorin leads to irregular collagen fibrils with highly variable diameters. Decorin is multifunctional and inactivates the profibrotic cytokines transforming growth factor β (TGF-β) and platelet-derived growth factor (PDGF) thereby inhibiting fibroblast activation and reducing tissue contraction. , Decorin also antagonizes and downregulates multiple profibrotic cell surface receptors, including epidermal growth factor receptor (EGFR), insulin-like growth factor 1 receptor (IGF-1R), and hepatocyte growth factor receptor (HGFR), leading to reduced migration and proliferation. In HSc, decorin is significantly reduced compared with normal skin, and as scars mature into NSc decorin production increases. Decorin upregulation reduces fibrosis. While decorin is downregulated in HSc, the proteoglycans biglycan and versican are upregulated significantly. , , Biglycan is highly similar to decorin and is believed to have originated as a gene duplication of decorin. Although biglycan is significantly upregulated in fibrosis, it does not compensate for the lack of decorin. , Versican is a large proteoglycan with 12 to 30 glycosaminoglycan (GAG) chains, which cause hyperhydration and the increased volume and turgor of HSc. Thus one of the mechanisms of action of pressure garments in reducing HSc is believed to be the reduction in hydration that results from external pressure on the water in scar tissues.
Cellular contributions to hypertrophic scar
Fibroblasts and their subtypes
Fibroblasts are the primary and most abundant cell type of connective tissue throughout the body and both the source and maintainers of ECM. A growing body of research demonstrates that there are many different subpopulations of fibroblasts with unique roles and responses to injury. , This heterogeneity of fibroblasts is even apparent in the dermis, where different fibroblast subtypes are located in different layers, with papillary (superficial) fibroblasts exhibiting significantly different behavior to that of reticular (deep) fibroblasts. , This heterogeneity also extends to myofibroblasts and HSc fibroblasts, which share many of the features of reticular (deep dermal) fibroblasts.
Dermal fibroblasts
Dermal fibroblasts can be divided into two major subpopulations with unique properties, including papillary (superficial) and reticular (deep) fibroblasts, which correspond to their depth and behavior within the dermis. , These fibroblast subpopulations differ both in their intrinsic properties and in their response to injury. Superficial dermal fibroblasts are regenerative in nature, whereas deep dermal fibroblasts are fibrotic. , Superficial dermal fibroblasts also produce ECM that is more supportive of keratinocyte viability and longevity than deep dermal fibroblasts. Ongoing research continues to delineate additional fibroblast subpopulations, including dermo-hypodermal junction fibroblasts, with unique behaviors and interactions with other cells.
Hypertrophic scar fibroblasts
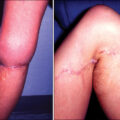
Fibroblasts of pathologic phenotypes can also be isolated from diseased tissues, such as fibrotic scars and cancer. Of the various fibroblast subtypes in normal skin, HSc fibroblasts most closely resemble the reticular (deep dermal) fibroblasts in vitro ( Table 44.1 ). This behavior is reflected in healing wounds of increasing depth as first studied by Dunkin et al., where a linear skin wound is created that goes from partial to full-thickness injuries. In these gradient wounds the superficial portion regenerates with virtually no scar, whereas the deeper portions heal with HSc , ( Fig. 44.2 ). Taken together, this suggests that HSc formation results from fibroblast behavior, which mirrors that of reticular fibroblast subtypes and could be the result of stimulation of reticular fibroblasts or the destruction of papillary fibroblasts, thus creating an imbalance. In a nude mouse model with human skin graft, the reticular fibroblasts closed wounds, whereas the papillary fibroblasts remodeled them, which is consistent with this pathogenic hypothesis. HSc fibroblasts may also reflect a chronic inflammatory phenotype, which can occur after prolonged stimulation with various cytokines, such as inteleukin 6 (IL-6).
Table 44.1
Superficial and Deep Dermal Fibroblasts Compared With Hypertrophic Scar Fibroblasts
| Superficial Dermal Fibroblasts | Deep Dermal Fibroblasts | Hypertrophic Scar Fibroblasts | |
|---|---|---|---|
| Collagen production | ↓︎ | ↑︎ | ↑︎ |
| Collagenase production | ↑︎ | ↓︎ | ↓︎ |
| Decorin production | ↑︎ | ↓︎ | ↓︎ |
| Transforming growth factor β production | ↓︎ | ↑︎ | ↑︎ |
| Connective tissue growth factor production | ↓︎ | ↑︎ | ↑︎ |
| Keratinocyte proliferation | ↑︎ | ↓︎ | ↓︎ |
| Capillary formation | ↑︎ | ↓︎ | ↓︎ |
Myofibroblasts
Myofibroblasts are terminally differentiated fibroblasts, characterized by intracellular α-smooth muscle actin (α-SMA), which occur in response to repetitive mechanical stresses, TGF-β stimulation, , granulocyte-macrophage colony-stimulating factor (GM-CSF), and other profibrotic factors. The expression of α-SMA is responsible for and proportional to the degree of ECM contraction. , Myofibroblasts are derived primarily from resident fibroblasts in the burn wound, but they can also be derived from multiple other cells types, such as epithelial and endothelial cells that undergo epithelial- or endothelial-mesenchymal transformation, mesenchymal stem cells, fibrocytes, and pericytes. Although TGF-β stimulates myofibroblast differentiation, this only occurs in the presence of fibronectin extra domain A (FN-EDA), production of which is also stimulated by TGF-β, suggesting a positive feedback loop. To some degree, myofibroblasts both create changes in ECM forces and respond to cyclic mechanical forces in ECM. When fibroblasts are cultured in gels and then stimulated with TGF-β, their expression of α-SMA is regulated by the compliance of the gel, and similarly culturing on stiff surfaces induces stress fiber formation, whereas soft surfaces do not. In physiologic wound healing, myofibroblasts ultimately undergo apoptosis, but in HSc they persist and contribute to the profibrotic environment ( Fig. 44.3 ).
Keratinocytes
As the body’s largest organ, skin is composed of two primary cell types: fibroblasts and keratinocytes, with keratinocytes serving to separate and maintain the internal body from the external environment. In wound healing, remodeling only begins once keratinocytes have reepithelialized the wound, and wounds where this takes more than 2 weeks are much more likely to form HSc. In skin equivalent models, keratinocytes downregulate the production of profibrotic TGF-β, connective tissue growth factor (CTGF) and collagen production by fibroblasts and increase collagen breakdown by matrix metalloproteinases (MMPs). However, keratinocytes can also contribute to the fibrosis of HSc through two mechanisms. First, keratinocyte phenotype alters in fibrotic conditions, leading to profibrotic keratinocytes, which can further stimulate profibrotic phenotypic changes in fibroblasts, creating a profibrotic feedback loop due in part to elevated PDGF production. Second, keratinocytes can undergo an epithelial to mesenchymal transition when stimulated by the profibrotic cytokine TGF-β, where they acquire a fibroblastoid phenotype. As a result, keratinocytes are not simply bystanders in HSc but active participants in sustaining HSc fibroblast phenotypes.
Systemic circulating cells
Wound healing and scarring are a complex response involving both resident cells in the injured tissue and systemic circulating cells (both platelets, and cells of immune system origin), which ultimately become involved in the local wound environment.
Fibrocytes
Fibrocytes are a CD14 + monocyte subpopulation that is collagen I and CD45 + or lymphocyte specific protein 1 (LSP1) + or CD34 + . , They are found in both normal wound healing and fibrosis such as HSc, and are attracted via a secondary lymphoid chemokine gradient, then differentiate following TGF-β stimulation. Fibrocytes contribute to ECM formation by differentiating into myofibroblasts and producing collagen and by modulating resident fibroblasts by secretion of TGF-β and CTGF. Fibrocytes can also act as antigen-presenting cells to prime naïve T cells, and express Toll-like receptors, thus acting as part of the innate immune system, thus serving as a link between the immune system and wound healing. Fibrocytes induce wound revascularization by secreting MMP-9, which promotes production of vascular endothelial growth factor (VEGF) and endothelial ingrowth. There also appear to be alternate fibrocyte subpopulations with antifibrotic properties, whose role in HSc is yet unknown, suggesting that the initial cytokines these bone marrow-derived cells encounter as they move from the circulatory system to wounds may ultimately determine their role in wound healing.
Immune system
The inflammatory phase of wound healing is intimately connected to the innate immune system, with mast cells, neutrophils, and macrophages playing an instrumental role. Neutrophils are one of the immune cells first recruited to wounds, and once present in wounds they stimulate keratinocytes and fibroblasts to proliferate. Increased infiltration of neutrophils and neutrophil extracellular traps are found in HSc and appear to stimulate myofibroblast differentiation. Furthermore, in normal wound healing, neutrophils undergo apoptosis and then phagocytosis by macrophages, which then downregulate inflammation, whereas wounds in which this does not occur undergo prolonged inflammation, which can lead to increased scarring. Macrophages stimulate wound keratinocytes and fibroblasts via immune cytokines IL-1, IL-6, and tumor necrosis factor α (TNF-α), as well as profibrotic cytokines TGF-β, PDGF, and IGF-1. Macrophages can be divided into two polarized groups: M1 and M2, with M2 polarization associated with HSc formation, , although the mechanism is unclear. The importance of polarization or type of immune response as opposed to degree of inflammation in HSc formation is crucial. HSc is highly infiltrated by CD4 + T-helper (T H ) cells, which can be divided into T H -1, T H -2, T H -17, and T-regulatory cells based on their cytokine profiles. In the T H 1-T H 2 axis, T H 1 cells are characterized by interferon γ (IFN-γ), IL-2, and TNF-β production, are involved in cell-mediated immunity and correspond to an antifibrotic profile, whereas T H 2 cells are characterized by IL-4, IL-5, and IL-10 production, are involved in antibody-mediated immunity and correspond to a profibrotic profile. , A T H 2 response is seen in human patients with over 25% total body surface area (TBSA) burns and human major trauma patients. In another longitudinal study of recovering burn patients, there was a predominant T H 2 response in peripheral blood mononuclear cells (PBMCs) and tissue, and this was significantly higher in those patients who developed HSc, whereas those with a T H 1 response developed NSc ( Fig. 44.4 ). A variety of mouse models confirm these findings and demonstrate that a T H 2 response displays more severe fibrosis, whereas modulation to a T H 1 response using antibodies or cytokines abrogates it. , ,
The role of cytokines in hypertrophic scar
Cytokines serve as the chemical signaling link between cells in paracrine signaling and cell self-modulation via feedback to the producing cell in autocrine signaling. Although there exists a tremendous number of cytokine signals, there are some well-studied cytokines that play key roles in fibrosis and HSc ( Fig. 44.5 ). The balance of profibrotic and antifibrotic cytokines is also crucial. In some cases, it is not purely the increase in profibrotic cytokines but also the decrease in antifibrotic cytokines that influences the ultimate formation of HSc.
Transforming growth factor β
TGF-β is perhaps the most studied and well-known prototypical profibrotic cytokine and is a member of a large superfamily of proteins that regulate embryonic development, chemotaxis, cell cycle, homeostasis, wound healing, immune cells differentiation, and a myriad of other processes. TGF-β is produced and secreted by fibroblasts bound to a latent TGF-β binding protein, which blocks its activity. This bond can be disrupted by mechanical forces or cleaved by MMP-2 or MMP-9 and plasmin (present in blood). Three isoforms are seen in mammals (TGF-β1, TGF-β2, TGF-β3) and are produced by most cells involved in wound healing, including platelets, T lymphocytes, macrophages, fibroblasts, keratinocytes, and endothelial cells. TGF-β is upregulated both locally in wounds and systemically in the circulation of burn patients. HSc fibroblasts produce more TGF-β than normal fibroblasts, and regenerative fetal fibroblasts produce scar in response to TGF-β stimulation. TGF-β directly alters fibroblast ECM production, upregulating collagen and downregulating decorin . TGF-β also induces fibroblasts to transform into myofibroblasts, epithelial cells to become fibroblastoid cells via epithelial to mesenchymal transdifferentiation, and reduces apoptosis of fibroblasts in healing wounds. TGF-β also upregulates or synergizes with a number of other cytokines in wound healing and thus plays a very broad role in the formation of HSc.
Connective tissue growth factor
CTGF (also called CCN2) is the prototypical CCN cytokine, with a structure of four linked regions with multiple binding domains that allow CTGF to serve multiple roles, including growth factor, TGF-β cofactor, and cellular-ECM integrin and proteoglycan binding interface. Although stimulation solely with TGF-β or CTGF induces a transient profibrotic response in fibroblasts, costimulation causes prolonged fibrosis, and CTGF levels in chronic fibrosis stay elevated even after TGF-β levels return to normal. This suggests that TGF-β initiates fibrosis and CTGF sustains it, and this pattern is seen in HSc, scleroderma, and other fibroses. , , CTGF stimulation is also involved in myofibroblast differentiation and angiogenesis and can form a profibrotic feedback loop with TGF-β. , Conversely, blocking CTGF activation with iloprost, antibodies, and siRNA all reduce fibrosis. ,
Platelet-derived growth factor
PDGF is delivered to wounds by platelets via injured capillaries and produced by resident fibroblasts. The four PDGF isoforms (A, B, C, D) form dimers and drive fibrogenesis through fibroblast proliferation, myofibroblast transformation, and increased ECM production. , These profibrotic hallmarks are seen in the role of PDGF in multiple fibroses, including scleroderma. PDGF also upregulates TGF-β receptors in fibroblasts, again leading to increased fibrosis. Conversely, blocking PDGF receptors or signal transduction reduces fibrosis in multiple animal models. ,
Insulin-like growth factor 1
In intack skin, IGF-1 is produced by epidermal sweat and sebaceous glands and is sequestered from dermal fibroblasts. Once a burn or other wound occurs, IGF-1 is released, and exposed fibroblasts then become activated; in cases of delayed reepithelialization, prolonged exposure and inflammation would result. IGF-1 serves as a regulator of ECM proteins, including glycosaminoglycan and collagen, , and upregulates TGF-β transcription by fibroblasts and downregulates collagenase activity, all shifting the balance of ECM remodeling toward fibrosis. These profibrotic effects are seen in many fibroses, including scleroderma and postburn HSc. ,
Interferons
The transition to HSc formation results not only from an increase in profibrotic growth factors but also an imbalance between pro- and antifibrotic cytokines. IFNs are used by immune cells in host defense activation and are divided into type I (IFN-α and IFN-β from leukocytes and fibroblasts) and type II (IFN-γ from T lymphocytes). The antifibrotic cytokines IFN-α2b and IFN-γ inhibit fibroblast proliferation while downregulating production of ECM collagen and fibronectin and reducing TGF-β production. In addition, IFN-α2b upregulates collagenase and reduces the tissue inhibitors of metalloproteinase 1 (TIMP-1), which promotes scar remodeling. IFN-α2b also reduces wound contraction and myofibroblast populations and promotes fibroblast apoptosis in later wound healing stages. , In a clinical trial, IFN-α2b therapy in burn patients normalized serum TGF-β levels and reduced both scar angiogenesis and volume. In trauma patients who formed keloids, PBMCs produced less IFN-α and IFN-γ, which also supports their antifibrotic role. This suggests imbalances between pro- and antifibrotic cytokines that promote HSc formation in burn patients.
Conclusion
HSc is an aberrant wound healing response, where excessive ECM is deposited beyond that required for wound closure, and profibrotic fibroblast activity continues despite reepithelialization. In common with other fibroproliferative disorders, the transition from normal fibroblast to HSc fibroblast phenotype can be initiated by one or more factors, such as TGF-β stimulation or repetitive mechanical stress. Ultimately this results in persistently activated fibroblasts that continue profibrotic activity despite the presence of signals, which ordinarily transition wound healing to remodeling phase and initiate myofibroblast apoptosis. This pathological phenotype is accompanied by abnormal ECM deposition, which differs from normal skin and NSc with disorganized, thin, irregular collagen bundles in nodules instead of the normal thick, organized fibers in a basket weave. The simultaneous decrease in key proteoglycans, such as decorin, promotes ECM disorganization and allows profibrotic cytokines in the dermis to go unchecked. The elevated profibrotic and decreased antifibrotic cytokine levels create an autocrine milieu that sustains fibrosis, and systemic fibrocytes and T H cells migrate to the wound and support the HSc fibroblast phenotype. Whether future therapies will prevent HSc fibroblast induction, increase HSc fibroblast apoptosis, cause HSc fibroblast transformation into remodeling fibroblasts, or involve some other mechanism is unknown. It is our continued hope, however, that the complex mechanisms of HSc formation will yield multiple targets for medical therapy, which will ultimately improve the quality of life of burn survivors and others who suffer with fibroproliferative diseases.
References
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


