Pathology.
The characteristic histopathological feature of BCC is a tumour containing cells with large, oval or elongated nuclei and relatively little cytoplasm. The nuclei closely resemble those of the epidermal basal cells, but the tumour cells themselves differ from basal cells by not showing an intercellular bridge on light microscopy. The nuclei in BCC commonly have a rather uniform and not an anaplastic appearance. Retraction artefact often surrounds the tumour itself on light microscopic sections [1].
Differential Diagnosis.
Differential diagnoses of BCC include SCC, malignant melanoma (pigmented basal cell carcinoma), melanocytic naevi (pigmented), Bowen disease (especially superficial basal cell carcinoma), psoriasis, eczema, sebaceous hyperplasia, molluscum contagiosum and adnexal tumours [1].
Treatment.
The most common therapeutic measure is excisional biopsy or curettage (with or without electrodesiccation) of the tumour. As the different clinical features of BCC generally correspond to the histopathological characteristics, the depth and width of the therapeutic excision should be carefully considered. As a further measure, 5-aminolaevulinic acid photodynamic therapy, topical imiquimod, topical fluorouracil, cryosurgery and excision Mohs micrographic surgery could be used, but radiation management of BCC in childhood should be avoided [1,4,6,10].
References
1 Wong CSM, Strange RC, Lear JT. Basal cell carcinoma. Br Med J 2003;327:794–8.
2 Belhadjali H, Moussa A, Yahia S et al. Simultaneous occurrence of two squamous cell carcinomas within a nevus sebaceous of Jadassohn in an 11-year-old girl. Pediatr Dermatol 2009;26:236–7.
3 Castori M, Castiglia D, Passarelli F, Paradisi M. Bazex–Dupré–Christol syndrome: an ectodermal dysplasia with skin appendage neoplasms. Eur J Med Genet 2009;52:250–5.
4 Goldberg LH, Firoz BF, Weiss GJ et al. Basal cell nevus syndrome: a brave new world. Arch Dermatol 2010;146:17–19.
5 Tilli CM, Van Steensel MA, Krekels GA, Neumann HA, Ramaekers FC. Molecular aetiology and pathogenesis of basal cell carcinoma. Br J Dermatol 2005;152:1108–24.
6 Oseroff AR, Shieh S, Frawley NP et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol 2005;141:60–7.
7 LeSueur BW, Silvis NG, Hansen RC. Basal cell carcinoma in children: report of 3 cases. Arch Dermatol 2000;136:370–2.
8 Fantini F, Gualdi G, Cimitan A, Giannetti A. Metastatic basal cell carcinoma with squamous differentiation: report of a case with response of cutaneous metastases to electrochemotherapy. Arch Dermatol 2008;144:1186–8.
9 Robinson JK, Dahiya M. Basal cell carcinoma with pulmonary and lymph node metastasis causing death. Arch Dermatol 2003;139:643–8.
10 Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol 2009;145:1431–8.
Squamous Cell Carcinoma (Epidermoid Carcinoma)
Definition.
Squamous cell carcinoma (SCC) is a malignant proliferation of the keratinocytes of the epidermis or its appendages which is rarely seen in children.
Aetiology and Pathogenesis.
Solar radiation is considered to be a frequent cause of most SCCs in fair-skinned people, and in those with xeroderma pigmentosum and albinism [1,2]. Furthermore, exposure to chemical carcinogens can sometimes be related; associated agents have included exposures to soot, paraffin, tar, mineral oil and anthracene. Occasionally, this tumour can develop secondarily in pre-existing dermatoses, such as on the scars of epidermolysis bullosa dystrophica, pansclerotic morphoea, old burn scars (Marjolin ulcer), X-ray scars, chronic leg ulcer, lupus vulgaris, acne conglobata, the sinuses of osteomyelitis, vaccination scars and arsenic keratoses. Other predisposing conditions are epidermodysplasia verruciformis, Fanconi anaemia, dyskeratosis congenita, congenital immune defects and immunosuppressive treatment, where SCC may develop during childhood and adolescence.
Several studies have shown links between specific aberrations in head and neck SCC and clinical outcome, loss of heterozygosity at 2q and 18q is commonly associated with poor prognosis, and loss of heterozygosity at 9p21 is associated with recurrence.
Clinical Features.
Squamous cell carcinoma commonly presents on sun-exposed areas (face, scalp, lips, back of the hand, and pinna). The clinical appearance is highly variable. The tumour may presents as a hard plaque or a papule, often with an opalescent feature, with telangiectasias, which develops a central ulceration covered with crusts. The borders become wide, elevated and indurated.
The risk of metastasis is low. Sites particularly associated with elevated risk for recurrence or metastasis includ: ear, lip/perioral, nose, periorbital and genitalia. The SCC in immunosuppressed patients or transplant patients or those with lymphoproliferative disorders is much more aggressive, regardless of their location [1–3].
Pathology.
Histopathology of SCC is characterized by irregular nests of epidermal cells that proliferate downwards and invade the dermis. The tumour cells show varying degrees of differentiation in the direction of keratinization. Dyskeratosis and horn pearls are found in some areas. The prognosis depends on the level of invasion and the degree of differentiation, according to the histological classification of SCC into grades I–IV. In grade I, most cells are differentiated and the tumour has not penetrated beyond the level of the eccrine glands. In grade IV most of the cells are atypical and undifferentiated. The greater the number of atypical cells and depth of invasion, the higher the degree of malignancy and the possibility of metastasis.
Differential Diagnosis.
The diagnosis of SCC is easy in most cases. Sometimes, differentiating it from keratoacanthoma, actinic keratosis, oral chancre of syphilis and pseudo-carcinomatous hyperplasia may pose difficulties.
Treatment.
The available treatment options for in situ SCC include cryotherapy, electrosurgery (curettage and electrodessication), topical treatment (5-fluorouracil or imiquimod), photodynamic therapy, radiation therapy, surgical excision and Mohs surgery.
In contrast, lesions that are more deeply invasive or have a higher risk of recurrence or metastasis generally require surgical excision (Mohs surgery). Radiation should be used with great caution in children. Systemic chemotherapy (cisplatin, carboplatinin, paclitaxel, docetaxel, 5-fluorouracil and methotrexate) is used exclusively for patients with metastatic disease. Recent data suggest that rapamycin may decrease the risk of developing skin cancer. Epidermal growth factor receptor is overexpressed in a number of malignant neoplasms (e.g. SCC). Cetuximab, a human chimeric antibody, showed efficacy in recurrent SCC in adults [1,4–6].
References
1 Agada FO, Patmore H, Alhamarneh O, Stafford ND, Greenman J. Genetic profile of head and neck squamous cell carcinoma: clinical implications. J Laryngol Otol 2009;123:266–72.
2 Cooper JZ, Brown MD. Special concern about squamous cell carcinoma of the scalp in organ transplant recipients. Arch Dermatol 2006;142:755–8.
3 Dimitroulakos J, Ye LY, Benzaquen M et al. Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin Cancer Res 2001;7:158–67.
4 Bauman JE, Eaton KD, Martins RG. Treatment of recurrent squamous cell carcinoma of the skin with cetuximab. Arch Dermatol 2007;143:889–92.
5 Kypson AP, Anderson CA, Lochmuller CM, Dizon K, Rodriguez E. Squamous cell carcinoma of the pericardium found during coronary bypass surgery of possible cutaneous origin. Ann Thorac Surg 2007;84:1735–6.
6 Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol 2009;145:1431–8.
Pilomatrix Carcinoma
Definition.
Pilomatrix carcinoma is a rare malignant counterpart of pilomatrixoma, a skin adnexal tumour originating from hair matrix cells. Pilomatrix carcinoma can arise as a solitary lesion de novo, or through transformation of a pilomatrixoma [1].
Clinical Features.
It usually presents as a firm solitary asymptomatic mass, deeply located in the dermis, with a predilection for the head and neck area, upper back and preauricular area. The tumour varies in size from 10 to 100 mm. Some cases have superficial ulcers that discharge fine white granules. Pilomatrix carcinoma is a neoplasm of low-grade malignancy that is characterized by locally aggressive, a tendency for recurrence but low risk of metastasis [1–3].
Pathology.
Histopathology of pilomatrix carcinoma is characterized by irregular islands of epithelial cells in a cellular stroma and multiple cystic spaces containing necrotic débris and focal calcifications. Gradual transition of pleomorphic basaloid cells to shadow cells is seen in many areas, with progressive calcification of the shadow cells. High mitotic rates and nuclear aberrations are observed in some areas of the tumour, prompting to a complete and carefully review of the entire surgical specimen for accurate diagnosis.
The immunohistochemical stain for the proliferation marker Ki-67 (MIB1) in the basaloid cells and expression of the cell proliferation associated antigen Ki 67 are found in the tumour. Beta-catenin mutations occur not only in pilomatricoma but also in pilomatrix carcinoma [1,3,4].
Differential Diagnosis.
The differential diagnosis includes pilomatrixoma, aggressive pilomatrixoma and BCC.
Treatment.
Excisional surgery with adequate margins is clearly indicated to prevent the risk of local recurrences. In recurrent pilomatrix carcinoma, no chemotherapy regimen has been demonstrated to provide local control or to arrest metastatic spread [3,5].
References
1 Aherne NJ, Fitzpatrick DA, Gibbons D, Armstrong JG. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol 2008;3:47.
2 Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol 2008;30:174–7.
3 Hardisson D, Linares MD, Cuevas-Santos J, Contreras F. Pilomatrix carcinoma: a clinicopathologic study of six cases and review of the literature. Am J Dermatopathol 2001;23:394–401.
4 Hassanein AM, Glanz SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol 2004;150:511–6.
5 Piel S, Denisjuk N, Schadendorf D, Dissemond J. Giant pilomatricoma: a benign tumor in an uncommon presentation. J Pediatr 2009;154:623.
Dermal Malignant Tumours
Rhabdomyosarcoma
Definition and Epidemiology.
Rhabdomyosarcoma (RMS) is a primary malignancy in children and adolescents that arises from embryonic mesenchyme precursor of striated muscle. It is the most common soft-tissue sarcoma of childhood and adolescence and represents 7–8% of all malignant tumours of childhood. Around 65% of cases appear in children under 6 years of age; the ratio of males to females is 1.3–1.5 : 1. Primary cutaneous presentation is extremely unusual. Cutaneous metastasis of RMS, although rare, may be the first manifestation of the disease. Approximately 15% of children with RMS present with metastatic disease and their prognosis has not improved significantly over the last 15 years, despite changes in therapy [1,2].
Aetiology and Pathogenesis.
Most cases of RMS occur in a sporadic manner but associations have been described with neurofibromatosis 1, Costello syndrome, congenital melanocytic naevi, Beckwith–Wiedeman syndrome and Li–Fraumeni syndrome.
The two histological subtypes of RMS, embryonal and alveolar, have distinct genetic alterations that may have a role in the pathogenesis of these tumours. Alveolar RMS typically has a chromosomal translocation at t(2;13) (q35;q14). This translocation fuses the PAX3 gene, believed to regulate transcription during early neuromuscular development, with the FKHR gene. It is believed that this fusion transcription factor may inappropriately activate transcription of genes that contribute to a transformed phenotype. The variant T(1;13) (p36;q14) fuses the PAX7 gene located on chromosome 1 with FKHR. Patients with tumours expressing the PAX–FKHR fusion tend to be younger and more likely to present with an extremity lesion, suggesting a distinct clinical phenotype. Polymerase chain reaction assays are available for confirmation of the diagnosis of alveolar RMS based on the presence of these fusion genes [1–5].
Clinical Features.
The clinical presentation of RMS will depend on the site of origin of the primary tumour, the age of the patient and the presence or absence of metastatic disease. The majority of symptoms will be related to compression of local structures and occasionally mild pain. There are no classic paraneoplastic syndromes associated with RMS.
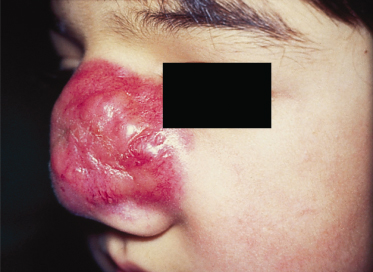
The most common sites in children are the head and neck region and the genitourinary tract, with only 20% involving the extremities. The disease can arise at any site and in any tissue in the body except bone. Primary cutaneous RMS develops mainly on the face as subcutaneous or protruding exophytic masses (Fig. 99.3). Orbital tumours produce proptosis and, occasionally, ophthalmoplegia. Those arising from parameningeal sites often produce nasal, ear or sinus obstruction with or without a mucopurulent or sanguinous discharge. Parameningeal tumours may extend into the cranium with resultant cranial nerve palsy and meningeal symptoms. When RMS is located on the trunk or limbs it is usually first noticed as a haematoma following trauma. The swelling persists or increases, and malignancy must be suspected.
Fig 99.3 Primary cutaneous rhabdomyosarcoma: bright red solid tumour infiltrating the nasal skin tissue.
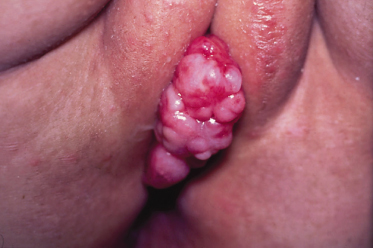
Vaginal tumours tend to occur in very young children and present with a mass or vaginal bleeding and discharge (Fig. 99.4).
Congenital alveolar RMS is a rare variant, characterized by multiple cutaneous metastases and an aggressive behaviour leading to early death [1,5].
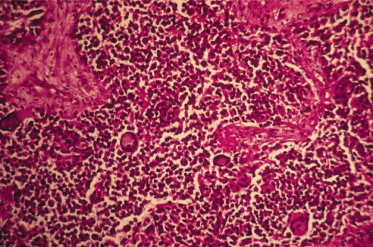
Pathology.
Rhabdomyosarcoma belongs to the group of ‘small round cell tumours’ (Fig. 99.5). Its myogenic origin can be assertained by immunohistochemical studies positive for muscle-specific actin, myogenin, desmin, vimentin, Myo D and Myo F4. There are four major histological subtypes:
1 Embryonal: the most common form in children;
2 Botryoid: a variant of the embryonal type in which tumour cells and an oedematous stroma protrudes into a body cavity (vagina, ear, nasopharynx);
3 Classic alveolar: these have cleft-like spaces between solid sheets of small cells resembling alveoli; and
4 Pleomorphic: rare in childhood.
Other recognized subtypes are spindle cell RMS, anaplastic RMS and solid-alveolar subtype RMS [1,2].
Diagnosis.
The diagnosis of RMS is usually made by direct open biopsy. There are no helpful serum markers. Patients with RMS require a complete work-up before definitive surgery: history/physical (height and weight); measure lesion (physical or imaging); complete blood count/differential/platelets; urinalysis; lytes, creatinine, calcium/phosphorus; alkaline phosphatase/lactate dehydrogenase/bilirubin, serum glucophosphatase; bone marrow biopsy/aspirate; chest radiograph; magnetic resonance imaging (MRI) or computed tomography (CT) of primary site; CT of chest; MRI or CT of head (for head tumours); bone scan; cerebral spinal fluid cytology (for parameningeal lesions) and electrocardiogram (ECG) or echocardiogram (selective) [1].
Differential Diagnosis.
Depending on the clinical appearance, RMS must be differentiated from haemolymphangioma, haemangioma, neuroblastoma, other soft-tissue sarcomas and haematoma. Radiography and CT provide clues about the extension and nature of the tumoural mass [2].
Treatment.
This is based on clinical staging according to primary tumour location and disease extension, and molecular biology. In group I tumours (localized disease), complete surgical excision is followed by chemotherapy. In group II (total gross resection with evidence of regional spread) and III tumours (incomplete resection with gross residual disease), local irradiation and multiple chemotherapy follows surgery. In group IV tumours (distant metastatic disease present at onset), systemic chemotherapy and irradiation are the best choices of treatment [1,2,6].
Prognosis.
Rapid diagnosis and recognition of this rare disorder will facilitate long-term survival. Currently, RMS has an overall survival of 70%. Depending on the age of the patient at the time of diagnosis, the histological type and the primary location, the tumour is classified into low, medium or high risk. Indicators of a poor prognosis are age under 1 year or over 10 years (Fig. 99.6), the alveolar histological type and primary location in the limbs [1,2,7].
References
1 Hayes-Jordan A, Andrassy R. Rhabdomyosarcoma in children. Curr Opin Pediatr 2009;21:373–8.
2 Lanoel A, Fain A, Cervini AB et al. Rhabdomyosarcoma with cutaneous presentation. Eur J Pediatr Dermatol 2009;19:8–14.
3 Lampe AK, Seymour G, Thompson PW, Toutain A, Lynch SA. Familial neurofibromatosis microdeletion syndrome complicated by rhabdomyosarcoma. Arch Dis Child 2002;87:444–5.
4 Hoang MP, Sinkre P, Albores-Saavedra J. Rhabdomyosarcoma arising in a congenital melanocytic nevus. Am J Dermatopathol 2002;24:26–9.
5 Grundy R, Anderson J, Gaze M et al. Congenital alveolar rhabdomyosarcoma: clinical and molecular distinction from alveolar rhabdomyosarcoma in older children. Cancer 2001;91:606–12.
6 Arndt CA, Hawkins DS, Meyer WH et al. Comparison of results of a pilot study of alternating vincristine/doxorubicin/cyclophosphamide and etoposide/ifosfamide with IRS-IV in intermediate risk rhabdomyosarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer 2008;50:33–6.
7 Meza JL, Anderson J, Pappo AS, Meyer WH; Children’s Oncology Group. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The Children’s Oncology Group. J Clin Oncol 2006;24:3844–51.
Other Soft-Tissue Sarcomas
The non-RMS soft-tissue sarcomas include tumours of different origins, which are extremely rare in childhood. They can occur as well as primary or as metastatic location to the skin and soft tissues [1]. Depending on the tissue type involved it is possible to observe the following:
1 Liposarcoma [2];
2 Fibrosarcoma, the most common in children under 1 year of age;
3 Epithelioid sarcoma [3];
4 Angiosarcoma [4];
5 Peripheral neuro-epithelioma or Ewing’s sarcoma [5];
6 Synovial sarcoma;
7 Alveolar soft part sarcoma, probably of myogenic origin; and
8 Leiomyosarcoma.
They commonly arise in the trunk or lower limbs. In all cases, surgical excision is the treatment of choice. Adjuvant chemotherapy or postoperative radiotherapy are used to treat residual lesions.
References
1 Wesche WA, Khare VK, Chesney TM, Jenkins JJ. Non-hematopoietic cutaneous metastases in children and adolescents: thirty years experience at St. Jude Children’s Research Hospital. J Cutan Pathol 2000;27:485–92.
2 Vocks E, Worret WI, Burgdorf WH. Myxoid liposarcoma in a 12-year-old girl. Pediatr Dermatol 2000;17:129–32.
3 Theunis A, André J, Larsimont D, Song M. Epithelioid sarcoma: a puzzling soft tissue neoplasm in a child. Dermatology 2000;200:179–80.
4 Lezama-del-Valle P, Gerald WL, Tsai J, Meyers P, La Quaglia MP. Malignant vascular tumors in young patients. Cancer 1998;83:1634–9.
5 Taylor GB, Chan YF. Subcutaneous primitive neuroectodermal tumour in the abdominal wall of a child: long-term survival after local excision. Pathology 2000;32:294–8.
Dermatofibrosarcoma Protuberans
Definition and Epidemiology.
Dermatofibrosarcoma protuberans (DFSP), first described by Darier, Ferrand and Hoffman, is a rare cutaneous tumour of fibrohistiocytic origin characterized by intermediate malignancy, locally aggressive growth with frequent local recurrence after treatment, but rare distant metastases with an estimated rate of 3.4–4.7%. The lungs are the most frequent site of metastasis, while lymph nodes are rarely involved. Dermatofibrosarcoma protuberans is typically diagnosed in young to middle-aged adults; only 6% of cases occur in paediatric patients [1,2].
Aetiology and Pathogenesis.
The mechanism by which DFSP arises is unknown. Trauma has been considered a possible aetiological factor, especially in childhood.
Specific cytogenetic abnormalities have been found in the majority of cases of DFSP. Supernumerary ring chromosome-containing sequence derived from chromosome 17 and 22 is the most consistent detectable cytogenetic change in adults. Reciprocal translocation t(17;22) (q22,q13) has been the most frequent (90%) abnormality in paediatric patients. Both of these rearrangements combine PDGFB (chromosome 22) and COL1A1 (chromosome 17) and could lead to increased production of PDGFb. It seems that autocrine PDGF-receptor stimulation contributes to neoplastic growth of these cells. Fusion of the COL1A1 gene with the PDGFB gene has also been detected in metastatic lesions of DFSP [1,3].
Clinical Features.
Dermatofibrosarcoma protuberans presents initially in childhood as a bluish discoloration or as a small and firm nodule or as plaque-like. The early-stage plaque can present different variants: an indurated flat plaque; a morphoea-like depressed sclerotic plaque; or an anetoderma-like depressed soft plaque. After a variable period of time, one or more nodules develop over the plaque. The colour ranges from brown to bluish-red, with a blue or red discoloration of the surrounding skin. They usually are asymptomatic and grow slowly. The lesion size normally ranges from 1 to 5 cm, tends to increase in dimension over the years and becomes larger if not treated. Dermatofibrosarcoma protuberans can occur in any part of the body, most often on the trunk, the proximal extremities and the head and neck region. However, in childhood the tumour seems to prefer the acral areas [2,3].
Pathology.
Histologically, the tumour is composed predominantly of spindle-shaped fibrohistiocytic cells arranged in short fascicles that interweave [1]. Mitotic figures are rare in most tumours. It is very often histologically indistinguishable from a low-grade fibrosarcoma or cellular fibroma occurring in the skin.
The fibrosarcomatous variant of DFSP represents an uncommon form. Histologically, the lesions show areas of typical low-grade DFSP adjacent to fibrosarcomatous areas. Immunohistochemically, tumour cells in DFSP areas stained positively for CD34, in contrast with the fibrosarcomatous areas that show only 50% of stained cells [1,4].
Differential Diagnosis.
Unfortunately, DFSP in children may be misdiagnosed, often as a vascular tumour, a malformation, morphoea, atrophoderma, atrophic scar and lipoatrophy, keloid and hypertrophic scars, but differs in the histological and immunohistochemical features.
The distinction between DFSP and dermatofibroma is sometimes difficult. Nestin is considered to be a useful marker in combination with CD34, factor XIIIa and CD163 in order to distinguish between DFSP and dermatofibroma [3–5].
Treatment.
The treatment of choice is wide local excision with a margin of 1.5–3 cm, including the underlying fascia. Mohs micrographic surgery has been used but this technique is not suitable for large lesions or for children.
Dermatofibrosarcoma protuberans is a radiosensitive tumour. Radiotherapy can be considered as a complement to surgery in cases of resection with committed margins. Excision and chemotherapy (vinblastine and oral methotrexate) have been used in the management of metastatic or recurring DFSP. Recent findings regarding the role of PDGFb and its receptor in the pathogenesis of DFSP have led investigators to target PDFG receptors as a novel treatment strategy for DFSP. Imatinib metylase, a specific tyrosine kinase inhibitor, has recently been reported to be effective in the setting of metastatic DFSP or local recurrence and large tumour [2,3,5,6].
References
1 Patel KU, Szabo SS, Hernandez VS et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol 2008;39:184–93.
2 Gerlini G, Mariotti G, Urso C et al. Dermatofibrosarcoma protuberans in childhood: two case reports and review of the literature. Pediatr Hematol Oncol 2008;25:559–66.
3 Jafarian F, McCuaig C, Kokta V, Laberge L, Ben Nejma B. Dermatofibrosarcoma protuberans in childhood and adolescence: report of eight patients. Pediatr Dermatol 2008;25:317–25.
4 Marque M, Bessis D, Pedetour F et al. Medallion-like dermal dendrocyte hamartoma: the main diagnostic pitfall is congenital atrophic dermatofibrosarcoma. Br J Dermatol 2009;160:190–3.
5 Mori T, Misago N, Yamamoto O, Toda S, Narisawa Y. Expression of nestin in dermatofibrosarcoma protuberans in comparison to dermatofibroma. J Dermatol 2008;35:419–25.
6 Bague S, Folpe AL. Dermatofibrosarcoma protuberans presenting as a subcutaneous mass: a clinicopathological study of 15 cases with exclusive or near-exclusive subcutaneous involvement. Am J Dermatopathol 2008;30:327–32.
Fibrosarcoma
Definition and Epidemiology.
Infantile fibrosarcoma is a rare soft tissue sarcoma arising at birth or in early infancy. It comprises less than 1% of all childhood tumours and approximately 10% of all sarcomas. Congenital fibrosarcoma is defined as tumour occurring within 3 months after birth. Congenital infantile fibrosarcoma is a low-grade myofibroblastic sarcoma. It is a borderline malignancy and has a more favourable prognosis than its adult counterpart. Infantile fibrosarcoma frequently recur locally, they have a relatively good prognosis and only rarely metastasize, and the lung is the preferential site for metastasis [1,2].
Clinical Features.
The tumour usually arises in the extremities, where it forms a hard irregular mass covered by normal, erythematous or purplish skin. The infantile form primarily affects the distal extremities, especially the ankle and foot, followed by the head and neck and then the trunk. The hand is also a site of predilection [3]. As it increases in size, ulceration and bleeding appear on the surface [2,4].
Pathology.
Fibrosarcoma is a tumour that presents many difficulties for histological diagnosis. The pattern of spindle-shaped cells, with a tendency to be arranged in fascicles or interlacing bands, is strongly suggestive of the diagnosis. Mitotic figures are common.
Cytogenetic studies of infantile fibrosarcomas have shown characteristic trisomies of chromosomes 8, 11, 17 and 20. A translocation has also been described between the TEL (ETV6) gene on 12p13 and the TRKC (NTRK3) gene on chromosome 15q25. This translocation has not been found in adult fibrosarcoma [1–4].
Differential Diagnosis.
The differential diagnosis includes congenital fibromatosis, myofibromatosis and RMS [2,3].
Treatment.
Standard treatment of infantile fibrosarcoma has been primarily surgical, with wide local excision. Histologically, free margins are the goal, without additional radiation and chemotherapy.
Infantile fibrosarcomas can be chemosensitive. Chemotherapy (vincristine, actinomycin-D, cyclophosphamide) has been used as a preoperative adjuvant to decrease the mass of the tumour before resection and in management of unresectable cases [1–4].
References
1 DeComas AM, Heinrich SD, Craver R. Infantile fibrosarcoma successfully treated with chemotherapy, with occurrence of calcifying aponeurotic fibroma and pleomorphic/spindled celled lipoma at the site 12 years later. J Pediatr Hematol Oncol 2009;31:448–52.
2 Gülhan B, Küpeli S, Yalçin B, Akyüz C, Büyükpamukçu M. An unusual presentation of infantile fibrosarcoma in a male newborn. Am J Perinatol 2009;26:331–3.
3 Yan AC, Chamlin SL, Liang MG et al. Congenital infantile fibrosarcoma: a masquerader of ulcerated hemangioma. Pediatr Dermatol 2006;23:330–4.
4 Russell H, Hicks MJ, Bertuch AA, Chintagumpala M. Infantile fibrosarcoma: clinical and histologic responses to cytotoxic chemotherapy. Pediatr Blood Cancer 2009;53:23–7.
Haemangiopericytoma/Solitary Fibrous Tumour
Definition.
Haemangiopericytoma (HPC) is an unusual vascular soft tissue tumour. In the most recent World Health Organization (WHO) classification of soft tissue sarcomas, the concept of HPC as a vascular pericyte-derived tumour was abandoned in favour of a fibroblastic cell of origin, thus placing HPC more closely with solitary fibrous tumour (SFT). It is much more common in adults; only 5–10% of these tumours occur in the paediatric population. When it develops in childhood, HPC is considered a heterogeneous entity that compromises two distinct clinical syndromes: adult HPC/SFT and infantile HPC. Infantile HPC, occurring during the first year of life, is characterized by a more benign course with responsiveness to chemotherapy and even tendency to spontaneous regression. In contrast, the behaviour of HPC in adolescence does not appear to differ from adult HPC. However, approximately 15–20% of HPC/SFT presents a more aggressive behaviour leading to development of local recurrences or distant metastases despite surgery [1,2].
Aetiology and Pathogenesis.
Exactly why this occurs is not entirely understood; however, studies have indicated that genetic alterations may have a role in formation of all soft tissue sarcomas. Although many different chromosomal rearrangements have been reported, it is clear that involvement of chromosome 12 is a frequent finding in HPC specimens. Specifically, translocations seem to be most common, with a breakpoint at 12q13-15 reported in multiple cases including paediatric cases. Only two inversions have been reported, one involving an X chromosome, the other involving chromosome 12 [inv (12) (q14q24)]. Both specimens with inversions belonged to adult patients [2].
Clinical Features.
Soft-tissue HPC is a firm, painless, slowly expanding mass that is often nodular and well localized (Fig. 99.7). The skin overlying the mass does not have any discoloration or redness to indicate its vascular origin because the capillaries are emptied of blood by compression of massive numbers of pericytes surrounding them.
The adult form is most common in the lower extremity but also occurs in the thorax, pelvis, retroperitoneum, orbit and other sites. Paediatric HPC particularly affects the limbs [1,3].
Pathology.
The infantile type, although histologically identical to the adult type, has a more benign clinical course. The reasons for these differences in the natural history of HPC are not well understood. Histologically, distinctive features of infantile HPC include immature cytology, multilobulated growth pattern, focal necrosis and mitotic activity in varying degrees. Capillaries are arranged in bands surrounded by cells with oval or spindle-shaped nuclei in a network of reticulum fibres. Additionally, some patients have a focal second tumour cell component, made of spindle-shaped myofibroblastic cells forming fascicles and micronodules.
Classically, immunohistochemical studies show expression of muscle-specific actin, smooth muscle actin, tropomyosin and CD34 and negativity for desmin and h-caldesmon, but this classic expression of markers is only present in 10–20% of HPC, with the majority expressing non-specific patterns [1,3].
Treatment.
Surgical excision is the definitive therapy for HPC. Chemotherapy has proved useful in a neoadjuvant setting for initially unresectable lesions and in cases where only a partial resection was possible or in presence of metastasis. Limited published data are available for the effectiveness of systemic chemotherapy in advanced HPC/SFT. Doxorubicin-based therapies have been used most frequently. The role of radiation therapy alone is controversial.
The rich vascular characteristics of HPC/SFT have been long recognized, and this observation has prompted the use of interferon-α in a few cases of advanced HPC/SFT based on the drug’s known anti-angiogenic properties.
Targeted therapy against the vascular endotelial growth factor (VEGF) pathway and other pathways involved in angiogenesis may provide a novel, exciting approach to treatment of HPC/SFT. Combination therapy with temozolomide and bevacizumab appears to provide clinical benefit with a low rate of major toxicities. Future prospective studies with other anti-angiogenic agents are necessary to investigate the biological mechanisms and the efficacy of these targeted agents in advanced HPC/SFT.
Infantile HPC is characterized by better clinical behaviour, with documented chemoresponsiveness and spontaneous regression, and requires a more conservative surgical approach [1,3–5].
References
1 Parka MS, Araujo DM. New insights into the hemangiopericytoma/solitary fibrous tumor spectrum of tumors. Curr Opin Oncol 2009;21:327–31.
2 Fletcher CD. The evolving classification of soft tissue tumours: an update based on the new WHO classification. Histopathology 2006;48:3–12.
3 Gowans LK, Bentz ML, DeSantes KB, Thompson KJ. Successful treatment of an infant with constitutional chromosomal abnormality and hemangiopericytoma with chemotherapy alone. J Pediatr Hematol Oncol 2007;29:409–11.
4 Lackner H, Urban C, Dornbusch HJ et al. Interferon alfa-2a in recurrent metastasic hemangiopericytoma. Med Pediatr Oncol 2003;40:192–4.
5 Perez EC, Brady CA, Luther W. Unusual non-epithelial tumors of the head and neck. In: Perez and Brady’s Principles and Practice of Radiation Oncology, 5th edn. Lippincott Williams & Wilkins, 2008:235–48.
Malignant Endovascular Papillary Angioendothelioma (Dabska Tumour)
Definition.
Dabska tumour is a rare low-grade angiosarcoma that often affects the skin of children. Maria Dabska originally described six patients in 1969 and named the disease malignant endovascular papillary angioendothelioma of the skin in childhood. Approximately 30 patients have been described worldwide.
This is a locally aggressive, low-grade neoplasm with metastatic potential. Thus, it has the ability to invade bone, muscle and fascia, producing local morbidity. It may also spread to regional lymph nodes and produce disseminated lung metastases with an unfavourable outcome [1].
Clinical Features.
Dabska tumour has no apparent predilection for any anatomical site. The head and extremities are commonly affected. Dabska tumour may also be seen on the palms, forearms, heels, knees, cheeks, temples, pinnae of the ears, neck, buttocks, abdominal skin and upper back.
Dabska tumour tends to be a slow-growing, painless intradermal nodule or tumour, violaceous, pink or bluish-black in coloration, 4–9 cm in diameter. An ulcerated large lesion with irregular, elevated and firm margins was seen in another case, arising over a large haemangioma [1–3].
Pathology.
Dabska tumour appears similar to cavernous lymphangiomas. A unique pattern is their papillary structure lined with atypical columnar endothelial cells. Mitotic figures are uncommon. Focal changes characteristic of retiform haemangioendothelioma may occasionally be observed. Immunoreactivity positive neoplastic cells demonstrate von Willebrand factor, CD31, CD34 and VEGFR-3 [1–3].
Differential Diagnosis.
Differential diagnosis includes acquired digital fibrokeratoma, angiolymphoid hyperplasia with eosinophilia, Kaposi sarcoma, reactive angioendotheliomatosis and angiosarcoma.
Treatment.
Wide local excision is the treatment of choice because of the risk of metastatic disease to the lymph nodes and lungs. Regional lymph node dissection may be indicated, especially if nodal involvement is detected [3].
References
1 Schwartz RA, Dabski C, Dabska M. The Dabska tumor: a thirty-year retrospect. Dermatology 2000;201:1–5.
2 Bhatia A, Nada R, Kumar Y, Menon P. Dabska tumor (endovascular papillary angioendothelioma) of testis: a case report with brief review of literature. Diagn Pathol 2006;1:12.
3 Emanuel PO, Lin R, Silver L et al. Dabska tumor arising in lymphangioma circumscriptum. J Cutan Pathol 2008;35:65–9.
Malignant tumours of neural crest and germ cell origin
Neuroblastoma
Definition and Epidemology.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


