Fig. 136.2 Rothmund–Thomson syndrome. Typical appearance of poikilodermatous changes mainly on the face, hands and legs.
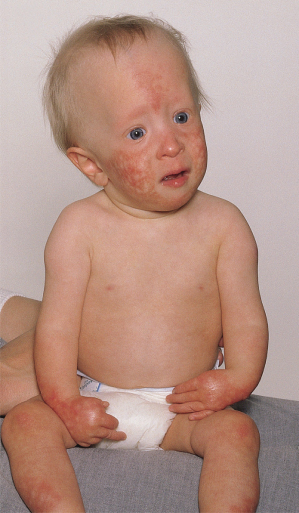
Transient photosensitivity has been reported in early childhood, but the typical distribution of inflammatory lesions on the face and backs of the hands may have been misinterpreted as photosensitivity. The occurrence of poikiloderma on the buttocks makes it unlikely that photosensitivity plays any part in the pathogenesis of this disorder.
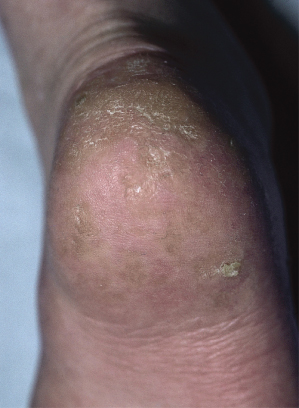
Keratoses are commonly found on the extremities of older children and adults (Fig. 136.3), and are considered premalignant [26].
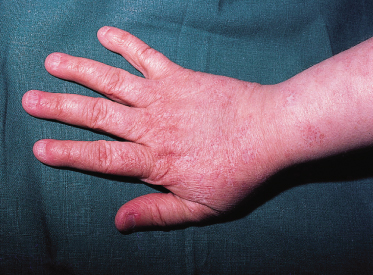
Sparse hair affecting particularly the scalp, eyebrows and eyelashes is commonly reported (see Fig. 136.2). Body hair may also be reduced. The nails are usually slow growing and hypotrophic (Fig. 136.4). A variety of dental anomalies has been reported [27]; none of them is characteristic.
The reported incidence of juvenile cataract has decreased with time, being 50% in Vennos’ [4] 1992 series and only 6% in Wang’s [6] 2001 series. Cataracts usually occur in all affected members of a sibship, and are generally of rapid onset, bilateral and of the subcapsular type; 73% develop before the age of 6 years [4]. A variety of other ocular abnormalities has been reported occasionally, including corneal atrophy, blue sclerae and iris dysgenesis [3,28].
Skeletal dysplasia is an important feature of Rothmund–Thomson syndrome [29]. Proportional short stature is common and may be severe. Radial ray defect is the most common specific skeletal abnormality, affecting 20% of patients [6], and presents as thumb hypoplasia or aplasia, with reduction or even complete absence of the radius. Some patients simply have an abnormal radial head, limiting elbow movement. The knees commonly show genu valgum and occasionally subluxation. Radiologically, metaphyseal chondrodysplasia, sclerosis and undertubulation of long bones are common, sometimes with a striking linear sclerosis affecting the femoral and tibial metaphyses, which may be a precursor to the development of osteosarcoma (see below). Midface hypoplasia with ‘saddle nose’ may be a marker for pituitary hypogonadism, having been reported in 10 out of 25 hypogonadal patients, but none out of 34 endocrinologically normal patients with Rothmund–Thomson syndrome [3].
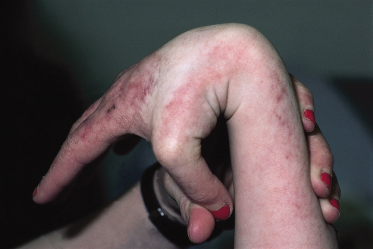
Acral hypermobility has been reported in several patients (Fig. 136.5). Multiple fractures, again mostly acral, and occasional blue sclerae also suggest a collagen defect but none has been identified [3].
About 40% of adult patients show delayed puberty or hypogonadism. Investigations have usually shown low or normal gonadotrophins suggestive of a primary pituitary defect. This may be structurally related to the midface hypoplasia [3].
An association between osteosarcoma and Rothmund–Thomson syndrome (RTS) was first noted in 1990 [3]. The clinical features of sporadic osteosarcomas and those associated with Rothmund–Thomson syndrome are similar except that the latter presents earlier [30]. Wang et al. [6] reported osteosarcoma in 13 out of a series of 41 patients (32%). Twelve published cases were reviewed in 1996 [31], and a further 12 patients were reviewed in 2007 [30], in whom osteosarcoma was the presenting feature of RTS in five. Ages at presentation of osteosarcoma ranged from 4 to 20 years (median 10), and in 75% of patients the tumour was in the tibia or distal femur. Many patients had previously abnormal bone with metaphyseal dysplasia and prominent longitudinal trabeculae. Three of 24 patients developed a second primary tumour. Disease-free survival was 83% at 5 years and 63% at 10 years [30]. Osteosarcoma in RTS patients shows abnormal sensitivity to chemotherapy, particularly doxorubicin, with prolonged myelosuppression and severe mucosal ulceration, but chemotherapy-related toxicity is not as severe in Rothmund–Thomson syndrome as in other DNA repair disorders [30]. Wang reported truncating mutations in RECQL4 in 23 out of 33 patients with Rothmund–Thomson syndrome and in all 11 of those with associated osteosarcoma [8]. RECQL4 is also one of several genes associated with sporadic osteosarcoma [32].
The other malignancy associated with Rothmund–Thomson syndrome is squamous carcinoma of the skin, which tends to occur on the distal extremities [33], sometimes at the site of acral keratoses. One 63-year-old man with RTS suffered several non-melanoma skin cancers [5]. By 2008 there were 61 patients with Rothmund-Thomson syndrome reported in the literature who had developed at least one malignancy, the most common being osteosarcoma (38 cases); there were three cases of squamous carcinoma of the tongue. Myelodysplasia [34] and amelanotic melanoma [35] have each once been reported in RTS as have several other malignancies.
Patients with Rothmund–Thomson syndrome are generally of normal intelligence, with no particular neurological problems.
Prognosis.
The increased risk of malignancy shortens life expectancy.
Differential Diagnosis.
Radial hypoplasia in a child too young to have developed the characteristic skin changes may be misdiagnosed as Fanconi syndrome or Holt–Oram syndrome [36]. The initial inflammation on light-exposed areas may be mistaken for photosensitivity. Blistering at the onset of poikiloderma may suggest Weary–Kindler syndrome (Fig.136.6). Later, the atrophic poikilodermatous skin and telangiectasia can mimic the premature ageing syndromes and Bloom syndrome respectively. Clericuzio-type poikiloderma with neutropenia is distinct from Rothmund–Thomson syndrome [37], while RAPADILINO and Baller–Gerold syndromes are allelic variants (see above). Both of these autosomal recessive syndromes, like RTS, are characterized by radial ray hypoplasia, facial dysmorphism and small stature, but are not as strongly associated with premature ageing or cancer, and hair and skin are usually normal [13]. Diarrhoea is a particular feature of RAPADILINO syndrome, and craniosynostosis of Baller–Gerold syndrome [38].
Treatment.
Treatment is largely symptomatic. Retinoids may improve the keratotic lesions [39]. It should be noted that chemotherapy for osteosarcoma has been associated with prolonged myelosuppression and severe mucosal ulceration [30]. An infant with Rothmund–Thomson syndrome and coincidental combined immunodeficiency was treated successfully with umbilical cord blood stem cell transplantation [40] but did not show any change in his poikiloderma or other features of RTS post transplant.
References
1 Rothmund A. Uber Kataract in Verbindung mit einer eigentumliche Haut Degenerationen. Arch Ophthal 1868;14:159–82.
2 Thomson MS. Poikiloderma congenitale. Br J Dermatol 1936;48:221.
3 Moss C. Rothmund–Thomson syndrome: a report of two patients and a review of the literature. Br J Dermatol 1990;122:821–9.
4 Vennos EM, Collins M, James WD. Rothmund–Thomson syndrome: review of the world literature. J Am Acad Dermatol 1992;27:750–62.
5 Stinco G, Governatori G, Mattighello P, Patrone P. Multiple cutaneous neoplasms in a patient with Rothmund–Thomson syndrome: case report and published work review. J Dermatol 2008;35(3):154–61.
6 Wang LL, Levy ML, Lewis RA et al. Clinical manifestations in a cohort of 41 Rothmund–Thomson syndrome patients. Am J Med Genet 2001;102:11–17.
7 Cabral RE, Queille S, Bodemer C et al. Identification of new RECQL4 mutations in Caucasian Rothmund–Thomson patients and analysis of sensitivity to a wide range of genotoxic agents. Mutat Res 2008;643(1–2):41–7.
8 Wang LL, Gannavarapu A, Kozinetz CA et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund–Thomson syndrome. J Natl Cancer Inst 2003;95:669–74.
9 Hickson ID. RecQ helicases: caretakers of the genome. Nature Rev Cancer 2003;3:169–78.
10 Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci 2008;33(12):609–20.
11 Lindor NM, Furuichi Y, Kitao S et al. Rothmund–Thomson syndrome due to RECQ4 helicase mutations: report and clinical molecular comparisons with Bloom syndrome and Werner syndrome. Am J Med Genet 2000;90:223–8.
12 Sznajer Y, Siitonen HA, Roversi G et al. Atypical Rothmund–Thomson syndrome in a patient with compound heterozygous mutations in RECQL4 gene and phenotypic features in RECQL4 syndromes. Eur J Pediatr 2008;167(2):175–81.
13 Siitonen HA, Sotkasiira J, Biervliet M et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet 2009;17(2):151–8.
14 Wang LL, Worley K, Gannavarapu A et al. Intron-size constraint as a mutational mechanism in Rothmund–Thomson syndrome. Am J Hum Genet 2002;71:165–7.
15 Beghini A, Castorina P, Roversi G et al. RNA processing defects of the helicase gene RECQL4 in a compound heterozygote Rothmund–Thomson patient. Am J Med Genet 2003;120A:395–9.
16 Dietschy T, Shevelev I, Stagljar I. The molecular role of the Rothmund–Thomson-, RAPADILINO- and Baller–Gerold-gene product, RECQL4: recent progress. Cell Mol Life Sci 2007;64:796–802.
17 Kellermayer R, Siitonen HA, Hadzsiev K et al. A patient with Rothmund–Thomson syndrome and all features of RAPADILINO. Arch Dermatol 2005;141(5):617–20.
18 Jin W, Liu H, Zhang Y et al. Sensitivity of RECQL4-deficient fibroblasts from Rothmund–Thomson syndrome patients to genotoxic agents. Hum Genet 2008;123(6):643–53.
19 Smith PJ, Paterson MC. Enhanced radiosensitivity and defective DNA repair in cultured fibroblasts derived from Rothmund–Thomson patients. Mutat Res 1982;94:213–28.
20 Shinya A, Nishigori C, Moriwaki S et al. A case of Rothmund–Thomson syndrome with reduced DNA repair capacity. Arch Dermatol 1993;129:332–6.
21 Judge MR, Kilby A, Harper JI. Rothmund–Thomson syndrome and osteosarcoma. Br J Dermatol 1993;129:723–5.
22 Koch F, Santamouris C, Ulbrich F. Trisomie bei einem Patienten mit Rothmund Syndrome. Zeitschr Kinderheilk 1967;99:1–13.
23 Durand F, Castorina P, Morant C et al. Rothmund–Thomson syndrome, trisomy 8 mosaicism and RecQ4 gene mutation. Ann Dermatol Venereol 2002;129:892–5.
24 Miozzo M, Castorina P, Riva P et al. Chromosomal instability in fibroblasts and mesenchymal tumours from two sibs with Rothmund–Thomson syndrome. Int J Cancer 1998;77:504–10.
25 Orstavik KH, McFadden N, Hagelsteen J et al. Instability of lymphocyte chromosomes in a girl with Rothmund–Thomson syndrome. J Med Genet 1994;31:570–2.
26 Shuttleworth D, Marks R. Epidermal dysplasia and skeletal deformity in congenital poikiloderma (Rothmund–Thomson syndrome). Br J Dermatol 1987;117:377–84.
27 Roinioti TD, Stefanopoulos PK. Short root anomaly associated with Rothmund–Thomson syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007;103(1):e19–22.
28 Mak RK, Griffiths WA, Mellerio JE. An unusual patient with Rothmund–Thomson syndrome, porokeratosis and bilateral iris dysgenesis. Clin Exp Dermatol 2006;31(3):401–3.
29 Mehollin-Ray AR, Kozinetz CA, Schlesinger AE et al. Radiographic abnormalities in Rothmund–Thomson syndrome and genotype–phenotype correlation with RECQL4 mutation status. AJR 2008;191(2):W62–6.
30 Hicks MJ, Roth JR, Kozinetz CA, Wang LL. Clinicopathologic features of osteosarcoma in patients with Rothmund–Thomson syndrome. J Clin Oncol 2007;25(4):370–5.
31 Leonard A, Craft AW, Moss C et al. Osteogenic sarcoma in the Rothmund–Thomson syndrome. Med Pediatr Oncol 1996;26:249–53.
32 Maire G, Yoshimoto M, Chilton-MacNeill S et al. Recurrent RECQL4 imbalance and increased gene expression levels are associated with structural chromosomal instability in sporadic osteosarcoma. Neoplasia 2009;11(3):260–8.
33 Piquero-Casals J, Okubo AY, Nico MM. Rothmund–Thomson syndrome in three siblings and development of cutaneous squamous cell carcinoma. Pediatr Dermatol 2002;19:312–16.
34 Pianigini E, de Aloe G, Andreassi A et al. Rothmund–Thomson syndrome (Thomson type) and myelodysplasia. Pediatr Dermatol 2001;18:422–5.
35 Howell SM, Bray DW. Amelanotic melanoma in a patient with Rothmund–Thomson syndrome. Arch Dermatol 2008;144(3):416–17.
36 Moss C, Bacon CJ, Mueller RF. ‘Isolated’ radial ray defect may be due to Rothmund–Thomson syndrome. Clin Genet 1990;38:318–19.
37 Van Hove JL, Jaeken J, Proesmans M et al. Clericuzio type poikiloderma with neutropenia is distinct from Rothmund–Thomson syndrome. Am J Med Genet A 2005;132A(2):152–8.
38 Van Maldergem L, Verloes A, Lejeune L, Gillerot Y. The Baller–Gerold syndrome. J Med Genet 1992;29(4):266–8.
39 Shuttleworth D, Marks R. Congenital poikiloderma: treatment with etretinate. Br J Dermatol 1988;118:729–30.
40 Broom MA, Wang LL, Otta SK et al. Successful umbilical cord blood stem cell transplantation in a patient with Rothmund–Thomson syndrome and combined immunodeficiency. Clin Genet 2006;69(4):337–43.
Bloom Syndrome
Definition.
Bloom syndrome is a rare inherited disorder characterized by growth retardation, immunodeficiency, photosensitivity, predisposition to cancer, telangiectasia and pigmentary abnormalities.
History.
Bloom, a New York City dermatologist, first described this syndrome in 1966 [1], and contributed to a Bloom Syndrome Registry set up subsequently by Dr James German. The registry closed to new accessions in 1991 and the 165 catalogued patients, mostly Ashkenazi Jews, have provided an invaluable source of data. German observed the first case of leukaemia in a Bloom syndrome patient in 1963, and correctly deduced from the fact that this patient already had excessive chromosome breakage that this represented a significant association [2].
Pathogenesis and Genetics.
The first cellular abnormalities identified in Bloom syndrome were increased sensitivity to UV light [3] and increased sister chromatid exchange (SCE) [4]. SCE refers to exchange of material between pairs of chromosomes newly replicated from the same parent chromosome in mitosis: there is no loss of genetic material so it may be of little pathogenetic importance, but it is useful as a diagnostic test and may reflect increased mutagenicity. Other relevant findings include increased chromosomal breakage, susceptibility of Bloom syndrome cells to oxidative stress, the SCE rate being reduced by antioxidants, and expression of an activated c-myc oncogene in Bloom syndrome lymphoblastoid cells [5]. There is increased expression of a common fragile site at 5q31 [6]. The reduced accumulation of the p53 tumour suppressor protein following DNA damage [7], is not specific for Bloom syndrome [8]. DNA ligase 1 activity is also reduced [9].
Bloom syndrome is inherited as an autosomal recessive trait. Most patients are of Ashkenazi Jewish extraction, the gene frequency in this population being about 1:110 [10]. Quantitative SCE facilitated the mapping of Bloom syndrome to the long arm of chromosome 15 by McDaniel and Schultz, who carried out complementation studies using a Bloom syndrome cell line with a stable SCE rate 10-fold higher than that of normal, and micro-cell-mediated chromosome transfer [11]. They found that insertion of a normal chromosome 15 into Bloom syndrome cells dramatically reduced the SCE rate. The localization of Bloom syndrome to chromosome 15 was confirmed by two different techniques. Homozygosity mapping in patients with Bloom syndrome and consanguineous parents showed a common homozygous section at 15q26.1 in 25 out of 26 cases, strongly suggesting the presence of the recessive Bloom syndrome gene at that locus [12]. Disomy analysis in a single patient with Prader–Willi syndrome and Bloom syndrome showed that the distal part of the long arm of both chromosome 15s was derived from the mother (maternal isodisomy for distal 15q) so that the patient was homozygous for all genes in this region, which presumably includes the recessive Bloom syndrome gene [13]. The Bloom syndrome gene is tightly linked to (but distinct from) the FES oncogene, and linkage disequilibrium between these two loci in Ashkenazi Jews is strongly suggestive of a founder effect, i.e. all affected individuals inherited the mutation from a common ancestor [10].
In 1995, Ellis et al. [14] identified the Bloom syndrome gene as RECQL (known as BLM), which maps to 15q26.1 and mutations are now well documented [15]. In a 2007 report from the Bloom Syndrome Registry [16], 64 different mutations were identified in 125 of 134 patients, 54 premature terminations and 10 missense mutations; 19 of the 64 mutations were recurrent, i.e. shared by two or more persons. The RecQ family of proteins is important in maintaining genome stability (see Rothmund–Thomson syndrome). BLM encodes a helicase which unwinds complementary double strands and functions as part of a protein complex [17]. As well as the BLM helicase, the BLM complex includes four other known components: a topoisomerase Topo3α which unknots the two strands by inserting temporary nicks, RPA which binds to single-stranded DNA, RMI1 which binds to the other components of the BLM complex, and RMI2 which orchestrates the activity of the whole complex. RMI2 also binds to and helps free up stalled replication forks (structures formed as complementary DNA strands separate). The BLM complex regulates homologous recombination (see above), and corrects cross-overs between homologous double-stranded DNA sequences. Such cross-overs can cause loss of heterozygosity, contributing to cancer. The BLM complex associates with the Fanconi anaemia (FA) complex in a supercomplex known as BRAFT (BLM, RPM, FA and Topo3α). Interaction between BLM helicase and RAD51 recombinase contributes to breast cancer susceptibility [18]. BLM also contributes to B-cell immunity by ensuring proper development and function of the various B-cell subsets while counteracting lymphomagenesis [19].
An increase in chromosomal structural abnormalities in sperm has been reported in heterozygous carriers [20]. SCE rate is increased in cultured chorionic villous cells from Bloom syndrome fetuses [21]: this test has now been superseded by DNA diagnosis in pregnancies at risk.
Pathology.
The histology of telangiectatic skin is non-specific, with epidermal atrophy, telangiectasia and a mild perivascular inflammatory infiltrate. Immunologically, there is reduced immunoglobulin M (IgM), possibly due to a failure of maturation of IgM-bearing B-lymphocytes [19,22].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


