Although congenital hypothyroidism is an isolated problem in most patients, it has been reported in association with Alström syndrome [27], the CHARGE association (coloboma, heart disease, atresia choanae, retarded growth and retarded development and/or central nervous system (CNS) anomalies, genital hypoplasia and ear anomalies or deafness) [28], trisomy 4p [29], Beckwith–Wiedemann syndrome [30], cutis marmorata telangiectatica congenita [31], cutis laxa [32] and as part of the Young–Simpson syndrome (congenital hypothyroidism, mental retardation, blepharophimosis) [33].
Cutaneous Signs of Congenital Hypothyroidism.
Skin involvement is initially characterized by a cool and dry feel on palpation, pronounced cutis marmorata and a translucent pallor resembling alabaster [2]. The pallor results from the combined effect of anaemia, poor peripheral perfusion, prolonged neonatal jaundice, carotenaemia and accumulated GAGs. The accumulation of GAGs in the skin and tongue eventually results in thickening of the skin (myxoedema) and macroglossia. The thickened skin is non-pitting and has a doughy, boggy feel on palpation. The thickening is most prominent around the eyes, lips, supraclavicular fossae, hands and feet. The combination of thickened facial skin, protruding tongue, depressed nasal bridge and mild hypertelorism results in a characteristic facies. Other mucocutaneous findings in established cases include lustreless, slow-growing hair, slow-growing nails and delayed eruption of deciduous teeth.
Acquired Hypothyroidism
Acquired hypothyroidism is more common in females [34,35]. It usually presents with non-specific symptoms such as lethargy, constipation, cold intolerance, arthralgias and myalgias, or with specific problems such as an enlarged thyroid gland, umbilical hernia, poor linear growth, developmental delay, delayed dentition, poor school performance, delayed puberty or a neuropsychiatric illness [36–39]. Uncommon clinical manifestations include precocious puberty with multicystic ovaries [40], a polymyocitis-like syndrome with raised creatine phosphokinase (CPK) [41], pericardial tamponade [42], rhabdomyolysis with renal failure and pericardial effusion [43] and hyperprolactinaemia [44].
Acquired hypothyroidism has been reported in association with Kocher–Debre–Semelgaine syndrome [45], Wyck–Grumbuch syndrome [46], acanthosis nigricans and insulin resistance [47], acquired von Willebrand disease [48,49], precocious puberty [50], Schinzel–Giedion syndrome [51], allogeneic haemopoietic stem cell transplantation in children treated with fractionated total body irradiation [52], hepatic haemangioendothelioma [53,54], infants with glycogen storage disease type 1 [55], Angelman syndrome [56] and Sturge–Weber syndrome [57]. Hypothyroidism secondary to autoimmune thyroiditis can evolve rarely into hyperthyroidism [58] or patients can develop other autoimmune diseases [59].
Mucocutaneous Findings in Acquired Hypothyroidism.
The mucocutaneous findings will depend on the severity and duration of hypothyroidism at presentation [36,37]. Patients with well-established disease will have cool, dry skin, exhibiting a ‘yellow-tinged’ pallor. Myxoedematous changes are evident in the form of macroglossia and thickening of the skin on the face (‘expressionless facies’), over the supraclavicular fossae and around the hands and feet. The myxoedematous changes can be associated with purpura and poor wound healing. The lateral third of the eyebrows is characteristically lost and the scalp and body hair is sparse, brittle and lustreless in appearance. Paradoxically, some infants and children develop hypertrichosis on the back and upper arms involving dark, terminal hairs [60]. Nail growth is diminished and is associated with brittleness and ridging of the nail plates.
Investigations.
The clinical diagnosis of hypothyroidism is easily confirmed by measuring serum levels of free thyroxine and/or thyrotropin [1,2]. The thyrotropin level will be high in patients with primary hypothyroidism and low in patients with hypothyroidsm secondary to hypothalamopituitary dysfunction. Antithyroid antibodies may be found in neonates with transient hypothyroidism secondary to the transplacental transfer of maternal antibodies [14] and in infants and children with autoimmune thyroiditis [37].
Infants with congenital primary hypothyroidism require further evaluation (e.g. radio-isotope scan) to determine the presence and location of thyroid tissue [2]. Other investigative findings in patients with congenital hypothyroidism include absence of the long bone epiphyses [2] and pericardial effusions [61].
Prognosis.
Newborn screening programmes for congenital hypothyroidism have enabled the early introduction of thyroxine replacement therapy. This has resulted in normal intelligence in infants with mild thyroxine deficiency and a significant reduction in the degree of intellectual impairment in infants with severe thyroxine deficiency [62,63]. In addition to the resolution of those clinical features directly attributable to thyroxine deficiency (e.g. myxoedema), thyroxine replacement therapy can resolve features that are not directly attributable to thyroxine deficiency, e.g. hypertrichosis [60], acanthosis nigricans (AN) and insulin resistance (IR) [47], precocious puberty [40,48], ovarian cysts [40], rhabdomyolosis [43], myositis with raised CPK [41], pericardial tamponade [42], hyperprolactinaemia [44] and von Willebrand disease [48,49].
Differential Diagnosis.
The differential diagnosis of congenital hypothyroidism includes trisomy 21, gonadal dysgenesis (Turner syndrome), mucopolysaccharidoses and Beckwith–Wiedemann syndrome. Trisomy 21 and Turner syndrome are easily distinguished from congenital hypothyroidism by chromosomal studies. Infants with one of the mucopolysaccharidoses are distinguished by marked hepatosplenomegaly and normal thyroid function tests. Infants with Beckwith–Wiedemann syndrome have accelerated growth, an omphalocoele rather than an umbilical hernia, and normal thyroid function tests.
Management.
Congenital and acquired hypothyroidism are treated with L-thyroxine therapy. The dose will vary with the age and weight of the patient and the cause (e.g. the dose required will be greater in athyroid infants). The benefits of high-dose versus low-dose thyroxine therapy in the management of congenital hypothyroidism remain unclear [64]. Its administration should be supervised by an experienced practitioner. Care must be taken when treating congenital hypothyroidism because of the risk of inducing congestive cardiac disease in the early stages of treatment secondary to the mobilization of fluid from myxoedematous tissues.
References
1 Fisher A, Dussault JH, Foley TP et al. Screening for congenital hypothyroidism: results of screening 1 million North American infants. J Pediatr 1979;94:700–5.
2 Grant DB, Smith I, Fuggle PW et al. Congenital hypothyroidism detected by neonatal screening: relationship between biochemical severity and early clinical features. Arch Dis Child 1992;67:87–90.
3 Shanker SM, Menon PSN, Karmarker MG et al. Dysgenesis of the thyroid is the common type of childhood hypothyroidism in environmentally iodine deficient areas of North India. Acta Paediatr 1994;83:1047–51.
4 Park SM, Chatterjee VK. Genetics of hypothyroidism. J Med Genet 2005;42:378–89.
5 Takamatsu J, Nishikawa M, Horimoto M et al. Familial unresponsiveness to thyrotropin by autosomal recessive inheritance. J Clin Endocrinol Metab 1993;77:1569–73.
6 Isichei UP, Das SC, Egbuta JO. Central cretinism in four successive siblings. Postgrad Med J 1990;66:751–6.
7 Braunstein GD, Kohler PD. Endocrine manifestations of histiocytosis-X. Am J Pediatr Hematol Oncol 1981;3:67–75.
8 Lucky AW, Howley PM, Megyesi K et al. Endocrine studies in cystinosis: compensated primary hypothyroidism. J Pediatr 1977;91:204–10.
9 Jain R, lsaac RM, Gottschalk ME et al. Transient central hypothyroidism as a cause of failure to thrive in newborn infants. J Endocrinol Invest 1994;17:631–4.
10 Magee LA, Downer E, Sermer M et al. Pregnancy outcome after gestational exposure to amiodarone in Canada. Am J Obstet Gynecol 1995;172:1307–11.
11 Hanukoglu A, Curiel B, Berkowitz D et al. Hypothyroidism and dyshormonogenesis induced by D-pencillamine in children with Wilson’s disease and healthy infants born to a mother with Wilson’s disease. J Pediatr 2008;153:864–6.
12 Barakat M, Carson P, Hetherton AM et al. Hypothyroidism secondary to topical iodine treatment in infants with spina bifida. Acta Paediatr 1994;83:741–3.
13 Lee YS, Loke KY, Ng SC et al. Maternal thyrotoxicosis causing central hypothyroidism in infants. J Paediatr Child Health 2002;38:206–8.
14 Matsuura N, Yamada Y, Nohara Y et al. Familial neonatal transient hypothyroidism due to maternal TSH binding inhibitor immunoglobulin. N Engl J Med 1980;303:738–41.
15 Okamura K, Salo K, Ikenoue H et al. Primary hypothyroidism manifested in childhood with special reference to various types of reversible hypothyroidism. Eur J Endocrinol 1994;131:131–7.
16 Samuels MH, Ridgeway EC. Central hypothyroidism. Endocrinol Metab Clin North Am 1992;21:903–19.
17 De Zegher F, Vanderschueren-Lodeweyckx M, Heinrichs C et al. Thyroid dyshormonogenesis: severe hypothyroidism after normal neonatal thyroid stimulating hormone screening. Acta Paediatr 1992;81:274–6.
18 Vulsma J, Menzel G, Abbad FC et al. Iodine induced hypothyroidism in infants treated with continuous cyclic peritoneal dialysis. Lancet 1990;336:812.
19 Burnett A, Carney D, Mukhopadhyay S et al. Thyroid involvement with Langerhans cell histiocytosis in a 3-year-old male. Pediatr Blood Cancer 2008;50:726–7.
20 Gabrilove JL, Ludwig AW. The histogenesis of myxoedema. J Clin Endocrinol Metab 1957;17:925–32.
21 Toublanc JE. Comparison of epidemiological data on congenital hypothyroidism in Europe with those of other parts of the world. Hormone Res 1992;38:230–5.
22 Rendon-Macias ME, Morales-Garcia I, Huerta-Hernandez E et al. Birth prevalence of congenital hypothyroidism in Mexico. Pediatr Perinat Epidemiol 2008;22:478–85.
23 Lorey FW, Cunningham GC. Birth prevalence of primary congenital hypothyroidism by sex and ethnicity. Hum Biol 1992;64:531–8.
24 Tsai WY, Lee JS, Wang TR et al. Clinical characteristics of congenital hypothyroidism detected by neonatal screening. J Formos Med Assoc 1993;92:20–3.
25 Tarim OF, Yordam N. Congenital hypothyroidism in Turkey: a retrospective evaluation of 1000 cases. Turk J Pediatr 1992;34:197–202.
26 Tahirovic H, Toromanovic A. Clinical presentation of primary congenital hypothyroidism: experience before mass screening. Bosnian J Basic Med Sci 2005;5:26–9.
27 Charles SJ, Moore AT, Yates JRW et al. Alström syndrome: further evidence of autosomal recessive inheritance and endocrinological dysfunction. J Med Genet 1990;27:590–2.
28 Marin JF, Garcia B, Quintana A et al. The CHARGE association and athyrosis. J Med Genet 1991;28:207–8.
29 Ioan DM, Ghitan T. Trisomy 4p: a new case of congenital myxedema. Endocrinologie 1991;29:111–14.
30 Chien CH, Lee JS, Tsai WY et al. Wiedemann–Beckwith syndrome with congenital central hypothyroidism in one of monozygotic twins. J Formos Med Assoc 1990;89:132–6.
31 Pehr K, Moroz B. Cutis marmorata telangiectatica congenita: long-term follow-up, review of the literature and report of a case in conjunction with congenital hypothyroidism. Pediatr Dermatol 1993;10:6–11.
32 Koklu E, Gunes T, Ozturk MA et al. Cutis laxa associated with central hypothyroidism owing to isolated thyrotropin deficiency in a newborn. Pediatr Dermatol 2007;24:525–8.
33 Stagi S, Bindi G, Lapi E et al. Congenital hypothyroidism in Young–Simpson syndrome. J Pediatr Endocrinol 2008;21:1089–92.
34 Demirbilek H, Kandemir N, Gonc EN et al. Hashimoto’s thyroiditis in children and adolescents: a retrospective study on clinical, epidemiological and laboratory properties of the disease. J Pediatr Endocrinol 2007;20:1199–205.
35 De Vries L, Bulvik S, Phillip M. Chronic autoimmune thyroiditis in children and adolescents: at presentation and during long-term follow-up. Arch Dis Child 2009;94:33–7.
36 Dallas JS, Foley TP. Hypothyroidism. In: Lifshitz F (ed) Pediatric Endocrinology: A Clinical Guide, 2nd rev edn. New York: Dekker, 1990:478–93.
37 Foley TP, Abbassi V, Copeland KC et al. Brief report: hypothyroidism caused by chronic autoimmune thyroiditis in very young infants. N Engl J Med 1994;330:466–8.
38 Keenan GF, Ostrov BE, Goldsmith DP et al. Rheumatic symptoms associated with hypothyroidism in children. J Pediatr 1993;123:586–8.
39 Chalk JN. Psychosis in a 15-year-old hypothyroid girl: myxoedematous madness. Aust NZ J Psychiatr 1991;25:561–2.
40 Sanjeevaiah AR, Sanjay S, Deepak T et al. Precocious puberty and large multicystic ovaries in young girls with primary hypothyroidism. Endocr Pract 2007;13:652–5.
41 Sbrocchi AM, Chedeville G, Scuccimarri R et al. Pediatric hypothyroidism presenting with a polymyocitis-like syndrome and increased creatinine: report of three cases. J Pediatr Endocrinol 2008;21:89–92.
42 Shastry RM, Shastry CC. Primary hypothyroidism with pericardial tamponade. Ind J Pediatr 2007;74:580–1.
43 Galli-Tsinopoulou A, Stylianou C, Kokka P et al. Rhabdomyolysis, renal failure, pericardial effusion, and acquired von Willebrand disease resulting from hypothyroidism in a 10-year-old girl. Thyroid 2008;18:373–5.
44 Alves C, Alves AC. Primary hypothyroidism in a child simulating a prolactin-secreting adenoma. Childs Nerv Syst 2008;24:1505–8.
45 Najjar SS. Muscular hypertrophy in hypothyroid children: the Kocher–Debre–Semelaigne syndrome. A review of 23 cases. J Pediatr 1974;85:236–9.
46 Van Wyk JJ, Grumbach MM. Syndrome of precocious menstruation and galactorrhoea in juvenile hypothyroidism: an example of hormonal overlap in pituitary feedback. J Pediatr 1960;57:416–35.
47 Ober KP. Acanthosis nigricans and insulin resistance associated with hypothyroidism. Arch Dermatol 1985;121:229–31.
48 Bruggers CS, McElligott K, Rallison ML. Acquired von Willebrand disease in twins with autoimmune hypothyroidism: response to desmopressin and L-thyroxine therapy. J Pediatr 1994;125:911–13.
49 Manfredi E, van Zaane B, Gerdes VE et al. Hypothyroidism and acquired von Willebrand’s syndrome: a systematic review. Haemophilia 2008;14:423–33.
50 Bhattacharya M, Mitra A. Regression of precocious puberty in a child with hypothyroidism after thyroxine therapy. Indian Pediatr 1992;29:96–8.
51 Santos H, Cordeiro I, Medeira A et al. Schinzel–Giedion syndrome. A patient with hypothyroidism and diabetes insipidus. Genet Couns 1994;5:187–9.
52 Bailey HK, Kappy MS, Giller RH et al. Time-course and risk factors of hypothyroidism following allogeneic hematopoietic stem cell transplanatation (HSCT) in children conditioned with fractionated total body irradiation. Pediatr Blood Cancer 2008;51:405–9.
53 Kalpatthi R, Germak J, Mizelle K et al. Thyroid abnormalities in infantile hepatic hemangioendothelioma. Pediatr Blood Cancer 2007;49:1021–4.
54 Mouat F, Evans HM, Cutfield WS et al. Massive hemangioendothelioma and consumptive hypothyroidism. J Pediatr Endocrinol 2008;21:701–3.
55 Melis D, Pivonello R, Parenti G et al. Increased prevalence of thyroid autoimmunity and hypothyroidism in patients with glycogen storage disease type I. J Pediatr 2007;150:300–5.
56 Paprocka J, Jamroz E, Kalina M et al. Angelman syndrome and hypothyroidism: coincidence or unique correlation? Neuroendocrinol Lett 2007;28;545–6.
57 Comi AM, Bellamkonda S, Ferenc LM et al. Central hypothyroidism and Sturge–Weber syndrome. Pediatr Neurol 2008;39:58–62.
58 Maenpaa J. Hypothyroidism preceding hyperthyroid Graves’ disease in two children. Acta Endocrinol 1983;251(suppl):27–31.
59 Wuthrich RP. Pernicious anaemia, autoimmune hypothyroidism and rapidly progressive anti-GBM glomerulonephritis. Clin Nephrol 1994;42:404.
60 Perloff WH. Hirsutism – a manifestation of juvenile hypothyroidism. JAMA 1955;157:651–2.
61 Rondanini GF, de Panizza G, Bollati A et al. Congenital hypothyroidism and pericardial effusion. Horm Res 1991;35:41–4.
62 Tillotson SL, Fuggle PW, Smith I et al. Relation between biochemical severity and intelligence in early treated congenital hypothyroidism: a threshold effect. BMJ 1994;309:440–5.
63 Gruters A, Krude H. Update on the management of congenital hypothyroidism. Horm Res 2007;68(suppl 5):107–11.
64 Ng Sm, Anand D, Weindling AM. High versus low dose of initial thyroid hormone replacement for congenital hypothyroidism. Cochrane Database Syst Rev 2009;1:CD006972.
Hyperthyroidism
Pathogenesis.
Hyperthyroidism in the paediatric population is usually due to autoimmune thyroid disease in the form of Graves disease (diffuse toxic goitre) [1–3] or Hashimoto thyroiditis [1]. In both conditions, hypersecretion of thyroxine is due to stimulation of the thyrotropin receptor on the thyroid gland by circulating antibodies. Rarely, hyperthyroidism is due to an overactive thyroid nodule [4], a multinodular goitre in association with McCune–Albright syndrome [5], increased secretion of thyrotropin from a pituitary adenoma [6], hypersecretion of thyrotropin because of pituitary unresponsiveness to thyroxine [7], iodine exposure [8], excessive intake of thyroxine or a germline mutation in the thyroid-stimulating hormone (TSH) receptor gene resulting in constitutive stimulation of the thyroid gland [9,10]. Hyperthyroidism can manifest in newborns secondary to the transplacental transfer of antithyroid antibodies from a thyrotoxic mother [11].
Although most of the cutaneous findings can be directly attributed to the hypermetabolic state induced by the increased levels of thyroxine and tri-iodothyronine, the development of pretibial myxoedema in Graves disease does not correlate with elevated thyroxine levels. These patients have a circulating factor that stimulates GAG production by fibroblasts from the pretibial and orbital areas [12]. Fibroblasts from unaffected body parts do not respond to the circulating factor.
Pathology.
Pretibial myxoedema is characterized by mucinous deposits that separate collagen in the mid- and reticular dermis [13]. The mucinous deposits are acellular and are readily identified with mucin stains such as toluidine blue and Alcian blue.
Clinical Features.
Hyperthyroidism can develop at any age, including the newborn period [1–3,8,11,14]. Females are more commonly affected than males in a ratio of 3–5:1, and there is a family history of thyroid disease in 37–50% of cases [1–3,14,15].
The usual presentation of hyperthyroidism is with an obvious goitre or one or more of the following non-specific symptoms: restlessness, nervousness, emotional lability, weight loss despite increased appetite, palpitations, eye prominence and/or stare, hyperactivity with reduced attention span, tremor, excessive sweating, heat intolerance and fatigue [1–3,14,15]. Many of these symptoms will lead to deterioration in school performance. Uncommon presenting symptoms include weight gain, amenorrhoea, dyspnoea on exertion and/or at night, hair loss, diarrhoea, polyuria, enuresis and accelerated linear growth [1–3,14,15].
Examination reveals a palpable goitre in all patients, with an associated bruit in approximately 50% of patients [1,2,15]. A tremor is usually evident and may be associated with choreo-athetoid like movements of the upper limbs [2]. Cardiovascular examination reveals resting tachycardia, a flow murmur and a pulse pressure of over 50 mmHg in most patients [1–3,15]. Eye involvement occurs in 60% of cases [1,2,15] and approximately 66% of prepubertal patients will be above the 75th percentile for height at the time of diagnosis [1,14,15]. Eye involvement is usually mild and consists of one or more of the following: conjunctival injection, chemosis, lid fullness, lagophthalmos and increased lacrimation. Cutaneous findings are usually restricted to a flushed appearance of the skin and a warm, clammy feel on palpation. Uncommon skin findings include thinning of the scalp hair, vitiligo, onycholysis (particularly of the fourth fingernail) and hyperpigmentation [1–3,15].
Paediatric patients with Graves disease rarely exhibit the characteristic clinical triad of pretibial myxoedema, severe ophthalmopathy and thyroid acropathy [1,2,15]. Pretibial myxoedema occurs in fewer than 2% of cases and represents mucinous infiltration of the pretibial skin. Occasionally, other sites can be affected, such as the posterior calves, the thighs, arms, trunk and the dorsum of the feet. The infiltration is usually bilateral and manifests as non-pitting nodules or plaques that can be skin coloured or yellow, red or brown in colour. The surface characteristically exhibits dilated follicular orifices that convey a peau d’orange appearance. Severe ophthalmopathy usually develops in association with pretibial myxoedema. It is characterized by exophthalmos, diminished eye movements, lid retraction and lid lag. The exophthalmos and diminished eye movements are due to mucinous infiltration within the orbit and extraocular muscles. Thyroid acropathy consists of clubbing and enlargement of the distal extremities. The enlargement is due to a combination of soft tissue hypertrophy and subperiosteal periostosis in the diaphyseal region of the metacarpals, metatarsals, phalanges and distal long bones. It is extremely rare in children.
Investigations.
The clinical diagnosis of hyperthyroidism is easily confirmed by measuring free thyroxine and tri-iodothyronine levels in the serum. The TSH levels are depressed except in those patients whose disease is due to hypersecretion of thyrotropin [6,7]. Patients with Graves disease and Hashimoto thyroiditis will have circulating thyroid-stimulating antibodies. Radio-active iodine test will distinguish Graves disease (high uptake) from thyroiditis (low uptake). Further investigation is required if an overactive thyroid nodule or pituitary dysfunction is suspected.
Prognosis.
The non-specific symptoms and signs of hyperthyroidism will resolve when the thyroxine level returns to normal. Pretibial myxoedema, severe ophthalmopathy and thyroid acropathy usually persist despite reduction in thyroxine levels.
Differential Diagnosis.
The hyperthyroid child is often labelled as being anxious or suffering from a psychiatric disturbance. Pretibial myxoedema can be confused with scleromyxoedema and lichen amyloidosis. Both conditions can be distinguished on the basis of history, other physical findings and skin biopsy.
Treatment.
Patients with Graves disease can be treated with antithyroid drugs [1–3,14.15,16,17], subtotal thyroidectomy [16,18,19,20,21] and/or radio-active iodine [19,22]. Antithyroid drugs are used initially, and approximately 50% of patients will undergo disease remission [1–3,13,17]. If compliance is poor, side-effects develop to the medication or prolonged therapy fails to induce disease remission, the patient can be treated with subtotal thyroidectomy or radio-active iodine. Patients with Hashimoto thyroiditis are usually treated with β-blockers because of the self-limiting nature of the problem.
Pretibial myxoedema is a difficult problem to treat because of its lack of response to topical steroids, intralesional steroids and oral steroids. A combination of topical clobetasol and oral pentoxifylline [23] and a combination of shave excision and subcutaneous injections of octreotide (an insulin-like growth factor type 1 antagonist) [24] were reported to be of benefit. Although one report noted clinical and histological improvement with high-dose intravenous gammaglobulin therapy [25], another reported no response to high-dose intravenous gammaglobulin [26].
References
1 Vaidya VA, Bongiovanni AM, Parks JS et al. Twenty-two years experience in the medical management of juvenile thyrotoxicosis. Pediatrics 1974;54:565–70.
2 Barnes HV, Blizzard RM. Antithyroid drug therapy for toxic diffuse goitre (Graves’ disease). 30 years experience in children and adolescents. J Pediatr 1977;91:313–20.
3 Gorton C, Sadeghi-Nejad A, Senior B. Remission in children with hyperthyroidism treated with propylthiouracil. Am J Dis Child 1987;141:1084–6.
4 Mizukami Y, Michigishi T, Nonomura A et al. Autonomously functioning (hot) nodule of the thyroid gland. A clinical and histopathological study of 17 cases. Am J Clin Pathol 1994;101:29–35.
5 Hamilton CR Jr, Maloof F. Unusual types of hypothyroidism. Medicine (Balt) 1973;52:195–215.
6 Avramides A, Karapiperis A, Triantafyllidou E et al. TSH-secreting pituitary macroadenoma in an 11-year-old girl. Acta Paediatr 1992;81:1058–60.
7 Gershengorn MC, Weintraub BD. Thyrotropin induced hyperthyroidism caused by selective pituitary resistance to thyroid hormone. A new syndrome of inappropriate secretion of TSH. J Clin Invest 1975;56:633–45.
8 Bryant WP, Zimmerman D. Iodine induced hyperthyroidism in a newborn. Pediatrics 1995;95:434–6.
9 Alberti L, Proverbio MC, Costagliola S et al. A novel germline mutation in the TSH receptor gene causes non-autoimmune autosomal dominant hyperthyroidism. Eur J Endocrinol 2001;145:249–54.
10 Ferrara AM, Capalbo D, Rossi G et al. A new case of familial non-autoimmune hyperthryroidism caused by the M463V mutation in the TSH receptor with anticipation of the disease across generations: a possible role of iodine supplementation. Thyroid 2007;17:677–80.
11 Smallridge RC, Wartofsky L, Chopra IJ et al. Neonatal thyrotoxicosis: alterations in serum concentrations of LATS protector, T4, T3, reverse T3, and 3, 3′T2. J Pediatr 1978;93:118–20.
12 Cheung HS, Nicoloff JT, Kamiel MB et al. Stimulation of fibroblast biosynthetic activity by serum of patients with pretibial myxedema. J Invest Dermatol 1978;71:12–17.
13 Truhan AP, Roenigk HH Jr. The cutaneous mucinoses. J Am Acad Dermatol 1986;14:1–18.
14 Bhadada S, Bhansali A, Velayutham P et al. Juvenile hyperthyroidism: an experience. Ind Pediatr 2006;43:301–7.
15 Mokhashi MH, Desai U, Desai MP. Hyperthyroidism in children. Indian J Pediatr 2000;67:653–6.
16 Dotsch J, Siebler T, Hauffa BP et al. Diagnosis and management of juvenile hyperthyroidism in Germany: a retrospective multicenter study. J Pediatr Endocrinol Metab 2000;13:879–85.
17 Kaguelidou F, Alberti C, Castanet M et al. Predictors of autoimmune hyperthyroidism relapse in children after discontinuation of antithyroid drug treatment. J Clin Endocrinol Metab 2008;93:3817–26.
18 Desjardins JG. Treatment of hyperthyroidism in children. Can J Surg 1983;26:252–3.
19 Rivkees SA. The treatment of Grave’s disease in children. J Pediatr Endocrinol 2006;19:1095–111.
20 Moreno P, Gomez JM, Gomez N et al. Subtotal thyroidectomy: a reliable method to achieve euthyroidism in Grave’s disease. Prognostic factors. World J Surg 2006;30:1950–6.
21 Sugino K, Ito K, Nagahama M et al. Surgical management of Grave’s disease: 10-year prospective trial at a single institution. Endocr J 2008;55:161–7.
22 Clark JD, Gelfand MJ, Elgazzar AH. Iodine-131 therapy of hyperthyroidism in paediatric patients. J Nucl Med 1995;36:442–5.
23 Pineda AM, Tianco EA, Tan JB et al. Oral pentoxifylline and topical clobetasol propionate ointment in the treatment of pretibial myxoedema, with concomitant improvement of Grave’s ophthalmopathy. J Eur Acad Dermatol Venereol 2007;21:1441–3.
24 Felton J, Derrick EK, Price ML. Successful combined surgical and octreotide treatment of severe pretibial myxoedema reviewed after 9 years. Br J Dermatol 2003;148:825–6.
25 Antonelli A, Navarrane A, Palla R et al. Pretibial myxedema and high dose intravenous immunoglobulin treatment. Thyroid 1994;4:399–408.
26 Terheyden P, Kahaly GJ, Zillikens D et al. Lack of response of elephantiasic pretibial myxoedema to treatment with high-dose intravenous immunoglobulins. Clin Exp Dermatol 2003;28:224–6.
Disorders of the Adrenal Glands
Cushing Disease and Cushing Syndrome
Pathogenesis.
The Cushingoid phenotype results from the excessive effect of glucocorticoids on body tissues. Cushing disease refers to the clinical phenotype resulting from an overproduction of glucocorticoids by the adrenal cortex secondary to hypersecretion of adrenocorticotropic hormone (ACTH) from a pituitary adenoma [1,2]. Cushing syndrome refers to the clinical phenotype resulting from an overproduction of glucocorticoids by the adrenal cortex because of micronodular adrenal hyperplasia, an adrenal adenoma or an adrenal carcinoma or secondary to stimulation by ectopically produced ACTH [1,2] or in response to gastric inhibitory polypeptide (GIP) stimulation of adrenal cells aberrantly expressing receptors for GIP [3]. Ectopic ACTH production has been reported in children with thymic carcinoid [1], bronchial carcinoid [1,2], pancreatic tumour [4,5], and an ovarian steroid cell tumour [6]. Some of the features of Cushing syndrome can also occur with the overuse of oral steroids, potent topical steroids [7], intralesional steroids [7], inhaled steroids [8] and intranasal steroids [9].
Most of the symptoms and signs of Cushing disease and Cushing syndrome are due to the direct effect of glucocorticoids on various body tissues. Androgens may contribute to some of the cutaneous findings in patients with Cushing syndrome due to micronodular adrenal hyperplasia or an adrenal tumour. Hyperpigmentation in patients with Cushing disease is due to stimulation of melanocytes by ACTH and related peptides.
Clinical Features.
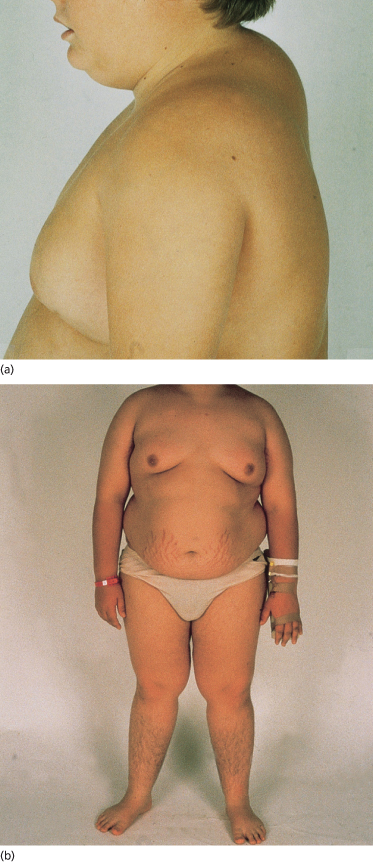
Cushing disease can occur at any age. Although there is no overall predilection for either sex [1,2,10] (Fig. 172.2), one study found a predeliction for males in the prepubertal age group [11]. Patients usually present with one or more of the following problems: weight gain, growth retardation in children and young adolescents, delayed sexual maturation in prepubertal patients, menstrual irregularities in postmenarchal females, fatigue, weakness, emotional lability and one or more of the characteristic skin findings [1,2,10,12]. The weight gain is associated with redistribution of body fat, resulting in a prominent moon face, buffalo hump and truncal obesity, with relative thinning of the arms and legs [1,2,10,12] (see Fig. 172.2). The characteristic skin findings are a plethoric facies, broad purple-coloured striae at the sites of skin tension (upper thighs, buttocks, trunk, shoulder girdle, breasts) (see Fig. 172.2b), skin fragility with poor wound healing, development of purpura with minimal skin trauma, hyperpigmentation, hypertrichosis (vellus-type hair) on the cheeks and forehead of the face, acanthosis nigricans and an acneiform eruption on the face and upper trunk [1,2]. The acneiform eruption represents either steroid folliculitis or exacerbation of teenage acne vulgaris. Other clinical findings include hypertension and osteopenia in the majority of patients.
Fig. 172.2 (a) Cushing disease exhibiting buffalo hump. (b) Cushing disease exhibiting truncal obesity and abdominal striae.
Courtesy of Dr Geoff Ambler, New Children’s Hospital, Sydney, Australia.
The clinical features of Cushing syndrome due to ectopic ACTH production are identical to those of Cushing disease. Cushing syndrome due to an adrenal tumour or micronodular adrenal hyperplasia is identical to Cushing disease, with the exception of androgen-mediated features such as hirsutism and premature adrenarche and the absence of ACTH-mediated hyperpigmentation. Steroid-induced Cushing syndrome is characterized by weight gain with redistribution of body fat, striae, skin fragility and an acneiform eruption [7–9].
Nodular adrenal hyperplasia can be associated with myxomatous tumours in the heart, skin and breast, centrofacial lentigines, testicular tumours (Sertoli cell tumour, Leydig cell tumour, adrenocortical rest tumour), pituitary adenomas and peripheral nerve tumours (schwannomas). This constellation of clinical features is referred to as the Carney complex (see below), a multisystem tumour syndrome inherited in an autosomal dominant fashion [13,14].
Investigations.
The clinical diagnosis of hypercortisolism is confirmed by measuring free cortisol levels in a 24-h urine specimen, measuring midnight sleeping plasma cortisol [15] and measuring the plasma cortisol levels after the low-dose dexametasone suppression test [1,2,11,12,16]. Patients with hypercortisolism will have elevated free cortisol levels in the urine and no decrease in plasma cortisol levels with low-dose dexametasone suppression [1,2,10,16]. Measurement of bedtime salivary cortisol appears to be as sensitive as urinary and plasma cortisol estimations [17]. ACTH-dependent disease can be distinguished from adrenal-mediated disease by measuring ACTH levels. If plasma ACTH levels are elevated, the high-dose dexametasone suppression test, the corticotropin-releasing hormone (CRH) stimulation test and inferior petrosal sinus sampling will distinguish pituitary and non-pituitary sources of ACTH [12,15]. If plasma ACTH levels are normal or depressed and the high-dose dexametasone suppression test fails to reduce plasma cortisol levels, imaging studies of the adrenal glands are required.
Differential Diagnosis.
Cushing disease needs to be excluded from physiological obesity and polycystic ovary disease. The measurement of serum cortisol and the dexametasone suppression test will distinguish Cushing disease and Cushing syndrome from physiological obesity. Polycystic ovary disease is distinguished by ultrasound of the ovaries and the presence of elevated luteinizing hormone (LH) and depressed follicle-stimulating hormone (FSH) levels.
Prognosis.
Provided the cause is identified and removed, the prognosis is good, with a gradual reduction in body weight, normal linear growth and the development of normal fertility [1,2,10].
Treatment.
Treatment depends on the underlying aetiology [1,2,10,12]. Pituitary tumours are removed surgically through the technique of trans-sphenoidal adenomectomy. Recurrent tumours can be surgically removed or treated with radiotherapy. Nodular adrenal hyperplasia and adrenal tumours are treated surgically. Nelson syndrome is a potential complication of adrenalectomy [10,18].
References
1 Magiakou MA, Mastorakos G, Oldfield EH et al. Cushing syndrome in children and adolescents. Presentation, diagnosis and therapy. N Engl J Med 1994;331:629–36.
2 Leinung MC, Zimmerman D. Cushing’s disease in children. Endocrinol Metab Clin North Am 1994;23:629–39.
3 Noordam C, Hermus AR, Pesman G et al. An adolescent with food-dependent Cushing syndrome secondary to ectopic expression of GIP receptor in unilateral adrenal adenoma. J Pediatr Endocrinol Metab 2002;15:853–60.
4 Kasperlik-Zauska AA, Jeske W, Cichocki A. Cushing’s syndrome in a 16-year-old girl due to ectopic ACTH precursor production from a pancreatic tumour. Clin Endocrinol 2001;55:558–9.
5 Illyes g, Luczay A, Benyo G et al. Cushing’s syndrome in a child with pancreatic acinar cell carcinoma. Endocr Pathol 2007;18:95–102.
6 Sawathiparnich P, Sitthinamsuwan P, Sanpakit K et al. Cushing’s syndrome caused by an ACTH-producing ovarian steroid cell tumour, NOS, in a prepubertal girl. Endocrinology 2009;35:132–35.
7 Curtis JA, Cormode E, Laski B et al. Endocrine complications of topical and intralesional corticosteroid therapy. Arch Dis Child 1982;57:204–7.
8 Priftis K, Everard ML, Milner AD. Unexpected side effects of inhaled steroids: a case report. Eur J Paediatr 1991;150:448–9.
9 Perry RJ, Findlay CA, Donaldson MD. Cushing syndrome, growth impairment and occult adrenal suppression associated with intranasal steroids. Arch Dis Child 2002;87:45–8.
10 Savage MO, Lienhardt A, Lebrethon MC et al. Cushing’s disease in childhood: presentation, investigation, treatment and long-term outcome. Hormone Res 2001;55(suppl 1):24–30.
11 Storr HL, Isidori AM, Monson JP et al. Prepubertal Cushing’s disease is more common in males, but there is no increase in severity at diagnosis. J Clin Endocrinol Metab 2004;89:3818–20.
12 Savage MO, Chan LF, Grossman AB et al. Work-up and management of pediatric Cushing’s syndrome. Curr Opin Endocrinol Diabet Obes 2008;15:346–51.
13 Carney JA, Hruska LS, Beauchamp BD et al. Dominant inheritance of the complex of myxomas, spotty pigmentation and endocrine over activity. Mayo Clin Proc 1986;61:165–72.
14 Carney JA, Gordon H, Carpenter PC et al. The complex of myxomas, spotty pigmentation and endocrine over activity. Medicine (Balt) 1985;64:270–83.
15 Batista DL, Riar J, Keil M et al. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics 2007;120:e575–86.
16 McLean M, Smith R. Cushing’s syndrome: how should we investigate in 1995? Med J Aust 1995;163:153–4.
17 Papanicolaou DA, Mullen N, Kyrou I et al. Nighttime salivary cortisol: a useful test for the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 2002;87:4515–21.
18 Thomas CG, Smith AT, Benson M et al. Nelson’s syndrome after Cushing’s disease in childhood: a continuing problem. Surgery 1984;96:1067–76.
Addison Disease
Pathogenesis.
Addison disease in the paediatric age group can result from reduced secretion of ACTH from the pituitary gland or from failure of the adrenal cortex to respond to ACTH. Reduced secretion of ACTH can be due to hypothalamopituitary axis dysfunction (e.g. pituitary tumour) or prolonged steroid therapy [1,2]. Failure of the adrenal cortex to respond to ACTH can be due to a variety of congenital and acquired conditions. Congenital problems include adrenal hypoplasia [3], congenital unresponsiveness to ACTH [4], adrenoleucodystrophy [5,6] and congenital adrenal hyperplasia [7]. Acquired causes include destruction of the adrenal gland as a result of infection or haemorrhage, surgical removal of the adrenal gland and an idiopathic cause. The idiopathic group exhibits a high incidence of circulating autoantibodies, particularly antibodies to the adrenal cortex cells, and most patients have a personal or family history of other autoimmune diseases [8]. The cumulative risk of developing Addison disease in the presence of adrenal cortex autoantibodies is approximately 50% [9]. The idiopathic group now accounts for most paediatric cases of Addison disease.
When glucocorticoid deficiency is associated with elevated ACTH levels, hyperpigmentation of the skin is a prominent feature. The pigmentation is due to stimulation of cutaneous melanocytes by ACTH and/or melanocyte-stimulating hormone (MSH) peptides.
Clinical Features.
The age of the patient at presentation will depend on the cause of the Addison disease. Patients with adrenal hypoplasia and congenital adrenal hyperplasia usually present shortly after birth. Patients with inadequate ACTH production, adrenal leucodystrophy and the idiopathic form can present at any age.
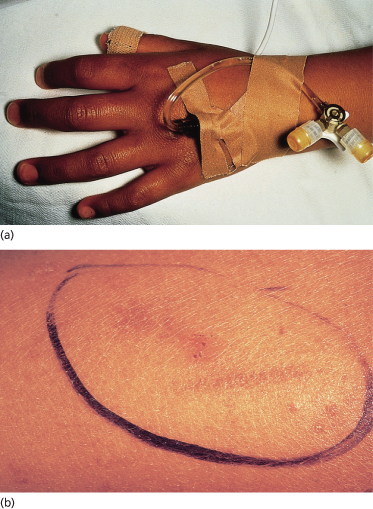
The usual presentation is with symptoms of lethargy, cyclic vomiting, hypotension and salt craving [7,8]. The onset of symptoms can be precipitated by an intercurrent illness [8]. Patients with gradual onset of the disease will exhibit hyperpigmentation of the skin and mucosal surfaces (gingivae, tongue, hard palate, buccal mucosa, vagina, anus) at the time of presentation [7,8]. The cutaneous pigmentation is diffuse, with accentuation on exposed areas (Fig. 172.3a), around new scars and over areas of pressure or friction (axillae, elbows, knees and perineum) (Fig. 172.3b). The development of mucosal pigmentation and palmoplantar crease pigmentation is restricted to Caucasians, as both are normal findings in black patients. The widespread hyperpigmentation is usually accompanied by darkening of the hair, darkening of existing melanocytic naevi and the appearance of longitudinal pigmented bands in the nail plate. Pubic and axillary hair may be lost in postpubertal females and vitiligo can be found in some patients.
Fig. 172.3 (a) Addison disease: hyperpigmentation on the dorsal aspect of the hand with accentuation over the knuckles. (b) Hyperpigmented scratch marks in a child with Addison disease.
Courtesy of Dr Geoff Ambler, New Children’s Hospital, Sydney, Australia.
Patients with adrenoleucodystrophy will eventually develop the typical neurological features of the disease [5,6]; notably, the diagnosis of Addison disease can precede the development of the neurological features of the disease by many years [6]. Female patients with congenital adrenal hyperplasia have ambiguous genitalia, which in the newborn period alerts the doctor to the possibility of adrenal insufficiency. The 3A syndrome refers to an association between glucocorticoid deficiency, alacrima, achalasia of the cardia, fissured palms, cutis anserina, autonomic nerve dysfunction and a variety of neurological abnormalities. Some affected patients also exhibit mineralocorticoid deficiency [10,11].
Investigations.
The clinical diagnosis of Addison disease is confirmed by measuring the plasma cortisol level and free cortisol levels in a 24-h urine collection [8]. Patients with Addison disease will have low plasma cortisol and urine free cortisol levels. Measurement of plasma ACTH levels and plasma cortisol levels following ACTH stimulation will distinguish adrenal failure from hypothalamopituitary dysfunction [8]. Most patients with the idiopathic form of Addison disease will have circulating antiadrenal antibodies [8]. Patients with adrenoleucodystrophy have raised plasma levels of hexacosanoic acid [6] and characteristic findings on magnetic resonance imaging of the brain [12].
Differential Diagnosis.
The early non-specific symptoms can be misdiagnosed as a wide variety of conditions. The hyperpigmentation is characteristic and readily distinguishes Addison disease from other conditions.
Prognosis.
All symptoms and signs will clear with appropriate glucocorticoid and mineralocorticoid replacement therapy. Patients with adrenoleucodystrophy will eventually develop neurological impairment.
Treatment.
Hydrocortisone and fludrocortisone are administered on a daily basis. The dose will need to be increased during periods of stress and patients with circulating autoantibodies will need to be regularly monitored for the development of other autoimmune diseases.
References
1 Curtis JA, Cormode E, Laski B et al. Endocrine complications of topical and intralesional corticosteroid therapy. Arch Dis Child 1982;57:204–7.
2 Molimard M, Girodet PO, Pollet C et al. Inhaled corticosteroids and adrenal insufficiency: prevalence and clinical presentation. Drug Safety 2008;31:769–74.
3 Sperling MA, Wolfsen AR, Fisher DA. Congenital adrenal hypoplasia: an isolated defect of organogenesis. J Pediatr 1973;82:444–9.
4 Kelch RP, Kaplan SL, Biglieri EG et al. Hereditary adrenocortical unresponsiveness to adrenocorticotropic hormone. J Pediatr 1972;81:726–36.
5 Davis LE, Snyder RD, Orth DN et al. Adrenoleukodystrophy and adrenomyeloneuropathy associated with partial adrenal insufficiency in three generations of a kindred. Am J Med 1979;66:342–7.
6 Sadhegi-Nejad A, Senior B. Adrenomyeloneuropathy presenting as Addison’s disease in childhood. N Engl J Med 1990;322:13–16.
7 Lim YJ, Batch JA, Warne GL. Adrenal 21-hydroxylase deficiency in childhood: 25 years experience. J Paediatr Child Health 1995;31:222–7.
8 Grant DB, Barnes ND, Moncrieff MW et al. Clinical presentation, growth and pubertal development in Addison’s disease. Arch Dis Child 1985;60:925–8.
9 Coco G, dal Pra C, Presotto F et al. Estimated risk for developing autoimmune Addison’s disease in patients with adrenal cortex autoantibodies. J Clin Endocrinol Metab 2006;1637–45.
10 Grant DB, Barnes ND, Dumic M et al. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome. Arch Dis Child 1993;68:779–82.
11 Toromanovic A, Tahirovic H, Milenkovic T et al. Clinical and molecular genetic findings in a 6-year-old Bosnian boy with triple A syndrome. Eur J Pediatr 2009;168:317–20.
12 Huckman MS, Wong PW, Sullivan T et al. Magnetic resonance imaging compared with computed tomography in adrenoleucodystrophy. Am J Dis Child 1986;140:1001–3.
Disorders of Sex Hormones
Hypogonadism
Pathogenesis.
Hypogonadism in females occurs with deficiency of gonadotropin secretion or inadequate production of sex hormones by the ovary or secondary to a virilizing disorder, hypothyroidism or hyperprolactinaemia [1]. Hypogonadism in males occurs with deficiency of gonadotropin secretion, inadequate androgen production by the testis, 5α-reductase deficiency or an androgen receptor defect [2].
Clinical Features.
The clinical features of hypogonadism in both sexes depend on the age of onset of the problem and the severity of the hormone deficiency [1–3]. Hypogonadism in a prepubertal female results in the failure of breast development (thelarche) and the onset of menarche (primary amenorrhoea). Pubic hair, axillary hair, body odour, sebum production and acne will still develop because of the unaltered production and secretion of adrenal androgens (adrenarche). The development of hypogonadism after the onset of puberty limits breast development and results in menstrual irregularities in the form of amenorrhoea, oligomenorrhoea or dysfunctional uterine bleeding. Linear growth is unaffected if growth hormone secretion is unaffected. Additional findings relate to the underlying cause of the hypogonadism (e.g. Turner syndrome).
In males, hypogonadism in utero results in abnormal (e.g. hypospadias) or ambiguous genitalia or infants who are phenotypically female. Prepubertal hypogonadism results in small testes, a small penis, lack of scrotal rugae, feminine fat distribution over the hips, face and chest, arm span 6 cm over height, eunuchoidal skeletal proportions (crown to pubis/pubis to floor ratio <1), decreased muscle mass and delayed bone age. The only notable skin findings are diminished body hair and a smooth, soft skin texture. If androgen deficiency is not corrected by the onset of adolescence, the skin will retain its soft texture and there will be no enlargement of the genitalia (testes and penis) or development of body and facial hair. Pubic and axillary hair may partially develop through the effect of adrenal androgens. Linear growth will continue if growth hormone secretion is normal, resulting in the eunuchoidal skeletal proportions. If androgen deficiency occurs after the onset of puberty, the clinical signs are more subtle. Established hair on the face, trunk, limbs, axillae and pubic area shows very little change, although the patient may notice that the interval between shaving gradually increases. The degree of skin oiliness will reduce, acne vulgaris will improve and often clear, and the skin texture becomes smooth, with fine wrinkles around the eyes and a decrease in the size of skin pores. Additional clinical findings relate to the underlying cause of the hypogonadism (e.g. Klinefelter syndrome).
Investigations.
Determining the cause of hypogonadism requires a detailed clinical assessment, determination of plasma gonadotropin (LH and FSH) levels, androgen levels, oestrogen levels and response of plasma gonadotropins to gonadotropin-releasing hormone. Imaging studies are needed if a tumour is suspected and chromosomal studies are required if the clinical features suggest Turner or Klinefelter syndrome.
Prognosis.
The prognosis is determined by the underlying cause.
Treatment.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


