Fig. 97.2 At the periphery of a myofibroma the individual cells are characterized by eosinophilic cytoplasm, round to oval dark nuclei and a grouping into nodular aggregates.
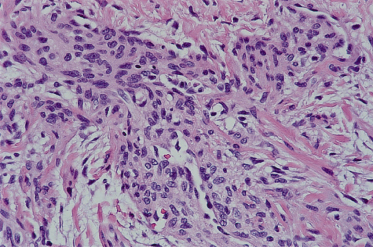
These features are the same in solitary and multiple lesions and so light microscopic distinction of these two different clinical forms of the disease is not possible following examination of a single lesion. A myofibromatous nodule differs from the infiltrative fibromatoses discussed below with regard to its borders; fibromatoses have infiltrative borders, whereas the myofibromas are relatively well circumscribed (although not actually encapsulated) on both gross and microscopic examination. Moreover, true fibromatoses produce a degree of stromal collagen not seen in myofibromas. In keeping with their myoid appearance, myofibroma cells show focal actin and vimentin positivity by immunohistochemistry; these lesions are desmin negative and S-100 protein negative.
Clinical Features.
Myofibromas are usually first appreciated when the parent or doctor notes the presence of a single (less often, multiple) subcutaneous or dermal nodule; these nodules may range in size from a few millimetres to 2 or 3 cm in their largest dimension. The nodules may be noted at birth or within the first few months of life; it is unusual for these lesions to develop after the first year or two of life (although occasional examples of myofibromas in older children and adults have been observed) [10]. The more superficially located lesions often have a bluish hue; deeply seated lesions are appreciated simply as firm to rubbery painless masses.
Two clinical patterns of disease may be identified: solitary disease (often subcutaneous), which is a self-limited process, and multifocal disease. Solitary lesions are particularly common in the head and neck region. In fact, one-third of such lesions occur in this region [2], solitary being more common than multifocal lesions. Patients with multifocal disease may develop lesions at a variety of sites, including the subcutis, skeleton, myocardium, gastrointestinal tract and lung. The skeleton may be involved in at least 50% of cases of multifocal disease [11]. These multifocal lesions may be quite numerous, ranging from a few dozen to perhaps 100 or more. The solitary lesions seem to be a more frequent occurrence amongst boys, whereas multifactorial disease appears to be more commonly seen in girls. Rarely, atrophic patches may be seen on neonates at sites of resolved ‘intrauterine’ lesions [12]. Among the multifocal (generalized) myofibromatoses there seem to be patients with a benign course and regression of the lesions [13], whereas other patients have a severe systemic disease with visceral involvement, leading to early adverse outcome [14]. The latter patients show significant clinical overlap with children affected by infantile systemic hyalinoses (see later in the chapter).
Differential Diagnosis.
Clinically, one may suspect any of a variety of tumours including neurofibroma, hyaline fibromatosis, vascular lesions such as haemangioma, haemangiopericytoma or sarcoma. Multicentric forms without spontaneous regression show overlap with infantile systemic hyalinosis and are probably manifestations of an autosomal recessive disorder [7,8]. Adequate biopsy material should distinguish these entities from myofibromatosis.
Prognosis.
Although the individual myofibromas themselves are not aggressive lesions, it is nevertheless important to distinguish those children with solitary lesions from those with multiple lesions [15,16]. Children with solitary lesions often see their lesions regress spontaneously (Fig. 97.3). Those patients with multifocal disease may be subdivided into two prognostic categories: those children with multifocal disease sparing the solid organs and those whose multifocal disease involves the viscera. It is the latter group – those children with visceral involvement – who may die of disease (particularly as a result of pulmonary or gastrointestinal tract involvement). Most deaths occur within the first week of life or no later than 4 months. Paradoxically, the myofibromas do not themselves exhibit malignant behaviour, in the sense of growing uncontrollably or metastasizing; rather, it is the sheer volume of these lesions in the vital organs that interferes with their function and so may lead to death.
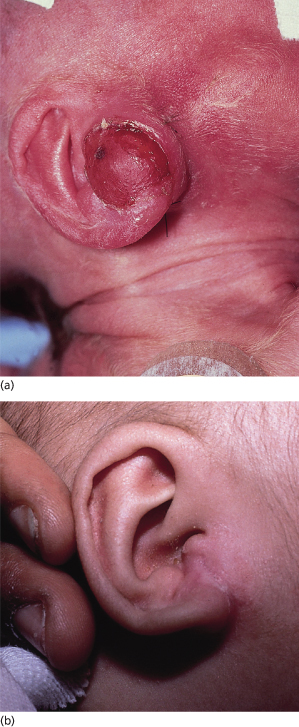
Fig. 97.3 (a) Firm erythematous tumour on the ear of a neonate typical of a myofibroma. (b) Same infant as in (a) at 6 months, with nearly complete spontaneous resolution of the myofibroma.
Treatment.
Ultimately, solitary myofibromas are best approached as benign lesions (with regard to their potential for uncontrolled growth), akin to hamartomas; this fact, coupled with recognition of the fact that many of these lesions regress over time, marks the myofibroma as a self-limited process. The solitary lesions are often excised surgically, and no more than 5–10% of these patients develop a local recurrence. Those children with multiple lesions are usually subjected to a biopsy for diagnosis and then observed, with many of their remaining lesions often regressing over time.
References
1 Chung ES, Enzinger FM. Infantile myofibromatosis. Cancer 1981;48:1807–18.
2 Hartig G, Koopman JC, Esciamado R. Infantile myofibromatosis: a commonly misdiagnosed entity. Otolaryngol Head Neck Surg 1993;109:753–7.
3 Dimmick JE, Wood WS. Congenital multiple fibromatosis. Am J Dermatopathol 1983;5:289–95.
4 Coffin CM, Pleiton KA, Angels S et al. Congenital generalized myofibromatosis: a disseminated angiocentric myofibromatosis. Pediatr Pathol Lab Med 1995;15:571–87.
5 Jennings TA, Sabetta J, Duray PH et al. Infantile myofibromatosis: evidence for an autosomal dominant disorder. Am J Surg Pathol 1984;8:529–38.
6 Ikediobi NI, Iyengar V, Hwang L et al. Infantile myofibromatosis: support for autosomal dominant inheritance. J Am Acad Dermatol 2003;49(Suppl.):148–50 (case reports).
7 Venencie PY, Bigel P, Desgruelles C et al. Infantile myofibromatosis: report of two cases in one family. Br J Dermatol 1987;117:255–9.
8 Narchi H. Four half-siblings with infantile myofibromatosis: a case for autosomal recessive inheritance. Clin Genet 2001;59:134–5.
9 Fukasawa Y, Ishikura H, Takada A et al. Massive apoptosis in infantile myofibromatosis: a putative mechanism of tumor regression. Am J Pathol 1994;144:480–5.
10 Stanford D, Rogers M. Dermatological presentations of infantile myofibromatosis: a review of 27 cases. Australas J Dermatol 2000;41:156–61.
11 Goldberg NS, Bauer BS, Kraus H, Crussi FG, Esterly NB. Infantile myofibromatosis: a review of clinicopathology with perspectives on new treatment choices. Pediatr Dermatol 1988;5:37–46.
12 Parker RK, Mallory SB, Baker GF. Infantile myofibromatosis. Pediatr Dermatol 1991;8:129–32.
13 Jenkins EA, Cawley MID. Infantile myofibromatosis: a cause of severe bone pain in a neonate. Br J Rheumatol 1993;32:849–52.
14 Familusi JB, Nottigde VA, Antia AU et al. Congenital generalized fibromatosis: an African case with gingival hypertrophy and other unusual features. Am J Dis Child 1976;130:1215–17.
15 Mentzel T, Calonje E, Nascimento AG et al. Infantile hemangiopericytoma versus infantile myofibromatosis: study of a series suggesting a continuous spectrum of infantile myofibroblastic lesions. Am J Surg Pathol 1994;18:922–30.
16 Variend S, Bax NMA, van Gorp J. Are infantile myofibromatosis, congenital fibrosarcoma and congenital haemangiopericytoma histogenetically related? Histopathology 1994;26:57–62.
Fibromatosis Colli
Definition.
Fibromatosis colli (synonyms: sternocleidomastoid tumour, congenital muscular fibromatosis) is a lesion seen exclusively in newborn infants and is marked by the development of a mass in the distal sternocleidomastoid muscle which often produces torticollis [1–3].
Aetiology.
In view of its exclusively congenital nature, many have thought that fibromatosis colli is likely to be related to some form of intrauterine trauma (perhaps sustained during the birth process itself) [3–5]. Although this may happen, cases have developed after uncomplicated spontaneous vaginal delivery, suggesting that factors other than birth trauma may be implicated. A clear-cut familial tendency has not been identified.
Pathology.
In its earliest phase of development, fibromatosis colli is marked by an infiltration of the skeletal muscle of the sternocleidomastoid muscle by a population of bland spindle cells, set in a myxoid background; neither cytological atypia nor mitotic activity is appreciable. As the lesion ages, its stroma becomes more heavily collagenized.
Clinical Features.
Fibromatosis colli usually comes to clinical attention in one of two ways: either the parent or the paediatrician notes a palpable mass in the vicinity of the lower portion of the sternocleidomastoid muscle (typically within the first few weeks after birth), or they observe torticollis or wryneck. This is, in the vast majority of instances, a unilateral process and appears more often in boys than in girls. The lesion is not fixed to the overlying skin, but rather seems to be associated with the adjacent muscle tissue. After a period of growth spanning only a few months, these masses usually stabilize or even regress.
Prognosis.
Most times, fibromatosis colli regresses spontaneously. It usually has an excellent prognosis. However, in a minority of patients, persistence of the mass may require surgical removal [6].
Differential Diagnosis.
Other causes of torticollis may enter into the clinical differential diagnosis (e.g. infection, trauma later in life and muscular spasm, which may all produce a tilting of the child’s head to one side or another, and so mimic fibromatosis colli). These are acquired lesions and so appear at a later age than fibromatosis colli.
Treatment.
As this lesion may regress spontaneously, treatment is often directed towards confirmation of the clinical impression of fibromatosis colli, by fine needle aspiration biopsy (favoured at present by many authors) [7] or by excisional biopsy. As an alternative, lesions clinically strongly suspected of being fibromatosis colli may be intermittently followed up to monitor for unexpected changes that might signal a more aggressive process [8]. Ultimately, surgical correction of any persisting deformity may be carried out in the minority of patients in whom such therapy is indicated [3,6].
References
1 McQueen WJ, Johnson JT, Edwards PA. Fibromatosis colli: a case report. Arch Otolaryngol Head Neck Surg 1980;88:49–51.
2 Thomsen JR, Koltai PJ. Sternomastoid tumor of infancy. Ann Otol Rhinol Laryngol 1989;98:955–9.
3 Bredenkarnp JK, Hoover LA, Berke GS et al. Congenital muscular torticollis: a spectrum of disease. Arch Otolaryngol Head Neck Surg 1990;116:212–16.
4 Tom LW, Handler SD, Wetmore RF et al. The sternocleidomastoid tumor of infancy. Int J Pediatr Pathol 1987;13:245–55.
5 Krugman ME, Canalis R, Konrad HR. The sternomastoid ‘tumor’ of infancy. J Otolaryngol 1976;5:523–9.
6 Cheng JC, Au AW. Infantile torticollis: a review of 624 cases. J Pediatr Orthop 1994;14:802–8.
7 Sharma S, Mishra K, Khanna G. Fibromatosis colli in infants: a cytologic study of eight cases. Acta Cytol 2003;47:359–62.
8 Ablin DS, Jain K, Howell L et al. Ultrasound and MR imaging of fibromatosis colli (sternomastoid tumor of infancy). Pediatr Radiol 1998;28:230–3.
Fibrous Hamartoma of Infancy
Definition and Aetiology.
The fibrous hamartoma of infancy (synonym: subdermal fibromatous tumour of infancy) appears to be a developmental anomaly, not a true neoplasm; a familial predilection for the development of these lesions has not been observed [1–5]. This anomaly has also been called lipofibromatosis because of the consistent presence of adipose tissue within the lesion.
Pathology.
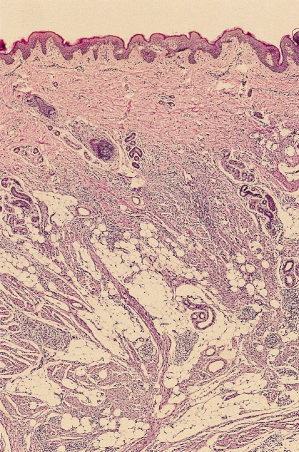
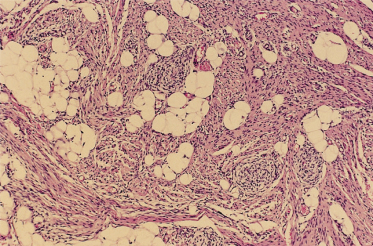
In its well-developed form, the fibrous hamartoma of infancy is a tripartite entity (referred to by some as an ‘organoid’ pattern); it contains areas of mature adipose tissue, bands of densely collagenized fibrous tissue and peculiar islands of immature cells that are disposed in a myxoid matrix (Fig. 97.4), all intimately admixed (Fig. 97.5) [6–8]. Although very occasional mitotic figures may be identified, they should not be prominent. The borders of these lesions may appear ill defined on microscopic examination, blending into the host tissues owing to the similarity of the adipose tissue to normal tissue.
Fig. 97.4 Fibrous hamartoma of infancy comprises three components: mature adipose tissue, aggregates of primitive cells and bands of fibrous tissue.
Clinical Features.
The fibrous hamartomas of infancy are painless nodular or plaque-like masses that appear to be centred in the subcutaneous tissues or the deep dermis (Fig. 97.6). Fibrous hamartomas of infancy are seen approximately twice as often in boys as in girls and are solitary lesions [9]. The lesion may be first noted either at birth or within the first 3–4 years of life; however, most lesions are diagnosed within the first 2 years. They are neither fixed to the skin nor to underlying deep structures. Commonly affected areas include the upper extremity and axilla, the neck and the lower extremity and groin region; the distribution therefore is central and not acral [1,2,4].
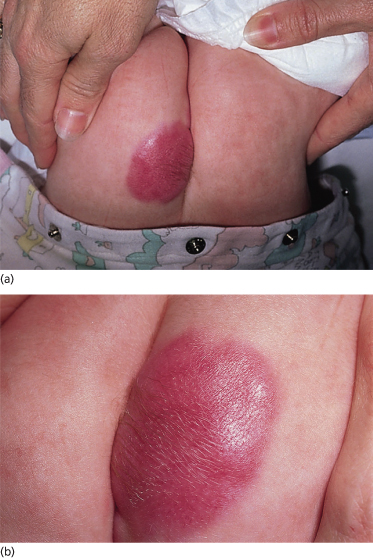
Prognosis.
Fibrous hamartomas of infancy may regress spontaneously. When surgically excised, they uncommonly recur. They are not premalignant lesions.
Differential Diagnosis.
If the presence of the immature areas is not appreciated (either because of sampling, or because they were simply overlooked) then fibrous hamartoma of infancy may be misinterpreted as a simple lipoma. The immature foci themselves may invite consideration of a malignant process; however, they are not marked by the same high rate of mitotic activity seen in malignant tumours. The spindled areas may resemble myoid or neural proliferations, but immunohistochemical studies will be of value in making this distinction (these fibrous areas in a fibrous hamartoma of infancy lack the desmin positivity of a smooth muscle tumour and they lack the S-100 protein positivity of a neural lesion). Finally, when attention is focused upon the fibrous component of this lesion, confusion with infantile fibromatosis is possible; however, identification of the immature mesenchymal component should permit distinction between these two entities.
Treatment.
Fibrous hamartomas of infancy are in most instances cured by a complete local excision. The recurrence rate following such therapy is less than 15% [10]. If a putative fibrous hamartoma of infancy recurs after attempted complete surgical excision, the original diagnostic material should be reviewed to exclude the possibility of a malignancy or other more aggressive process.
References
1 Enzinger FM. Fibrous hamartoma of infancy. Cancer 1965;18:241–8.
2 Pailer AS, Gonzliez-Crussi F, Sherman JO. Fibrous hamartomas of infancy: eight additional cases and a review of the literature. Arch Dermatol 1989;125:88–91.
3 Fletcher CD, Powell G, van Noorden S et al. Fibrous hamartoma of infancy: a histochemical and immunohistochemical study. Histopathology 1988;12:65–74.
4 Popek EJ, Montgomery EA, Fourcroy JL. Fibrous hamartoma of infancy in the genital region: findings in 15 cases. J Urol 1994;152:990–3.
5 Efem SE, Ekpo MD. Clinicopathological features of untreated fibrous hamartoma of infancy. J Clin Pathol 1993;46:522–4.
6 Greco MA, Schinella RA, Vuentin JC. Fibrous hamartoma of infancy: an ultrastructural study. Hum Pathol 1984;15:717–23.
7 Groisman G, Lichtig C. Fibrous hamartoma of infancy: an immunohistochemical and ultrastructural study. Hum Pathol 1991;22:914–18.
8 Michael M, Mukensnabi P, Chlumska A et al. Fibrous hamartoma of infancy: a study of eight cases with immunohistochemical and electron microscopical findings. Pathol Res Pract 1992;188:1049–53.
9 Dickey GE, Sotelo-Avila C. Fibrous hamartoma of infancy: current review. Pediatr Dev Pathol 1999;2:236–43.
10 Lee UT, Girvan DP, Armstrong RF. Fibrous hamartoma of infancy. J Pediatr Surg 1988;23:759–61.
Calcifying Fibrous Pseudo-Tumour
Definition and Aetiology.
The calcifying fibrous pseudo-tumour is a recently described entity that has only a limited potential for local recurrence. There is some debate as to its origin: some authors have suggested that it is a true neoplasm, whereas others believe that it represents a late stage (‘burnt-out’) inflammatory myofibroblastic tumour. To date, only a few dozen of these lesions have been described in the English language literature [1–4].
Pathology.
These lesions are reasonably well circumscribed; better circumscribed, for example, than the desmoid-type fibromatoses discussed below. A sclerotic collagenous stroma is a dominant feature of calcifying fibrous pseudo-tumours on low-power microscopy; on closer scrutiny, widely scattered spindle cells are found distributed throughout this collagenous stroma. A minimal chronic inflammatory cell infiltrate may also be scattered throughout this stroma. This lesion’s most characteristic microscopic feature, however, is the presence of calcified bodies (psammomatous calcifications), which are found within the mass (either throughout the lesion or as only a focal finding).
Clinical Features.
Calcifying fibrous pseudo-tumours may involve the subcutaneous tissues as well as deeper soft tissues; rare reports place these lesions in visceral locations. The typical patient is between 10 and 30 years of age. The majority of these lesions are located in the extremities; this does not seem to be a congenital process and a sex predilection has not been noted [2].
Prognosis.
The calcifying fibrous pseudo-tumour has an excellent prognosis. It is a slowly growing mass that is usually successfully treated by surgical excision. Local recurrence is rare and may occur several years after the initial surgical excision.
Differential Diagnosis.
Although clinical consideration might be given to a variety of fibrous proliferations (including desmoid-type fibromatosis, digital fibromatosis and nodular fasciitis), the calcifications are a distinctive feature whose presence, along with the distinctly low cellularity and relatively sharp circumscription from adjacent tissue, should suggest the diagnosis of calcifying fibrous pseudo-tumour.
Treatment.
Complete local excision seems to be the best approach to treatment, as only a minority of patients will experience local recurrences following such a procedure [2].
References
1 Rosenthal NS, Abdul-Karim FW. Childhood fibrous tumor with psammona bodies. Arch Pathol Lab Med 1988;112:798–800.
2 Fetsch JF, Montgomery EA, Meis JM. Calcifying fibrous pseudotumor. Am J Surg Pathol 1993;17:502–8.
3 Hill KA, Gonzalez-Crussi F, Chou PM. Calcifying fibrous pseudotumor versus inflammatory myofibroblastic tumor: a histological and immunohistochemical comparison. Mod Pathol 2001;14:784–90.
4 Nascimento AF, Ruiz R, Hornick JL et al. Calcifying fibrous ‘pseudotumor’: clinicopathologic study of 15 cases and analysis of its relationship to inflammatory myofibroblastic tumor. Int J Surg Pathol 2002;10:189–96.
Nodular Fasciitis
Definition and Aetiology.
Nodular fasciitis (synonym: subcutaneous pseudo-sarcomatous fibromatosis) is a reactive myofibroblastic proliferation. It has not usually been included in standard classification schemes of the fibromatoses of childhood, but its light microscopic appearance may overlap with that of the fibromatoses and so it is considered here [1–6]. Although some lesions of nodular fasciitis have been associated with a prior history of trauma, the majority have not; the lesion, nevertheless, suggests a reactive proliferation on light microscopy, and so a suspicion persists that it might in some way be related to some inflammatory or traumatic stimulus [4,7,8].
Pathology.
The essential light microscopic features include a loose proliferation of spindle cells set in a myxoid stroma, scattered foci of haemorrhage and scattered lymphocytes. Mitotic figures are often readily identifiable but are not, however, atypical appearing.
Uncommonly, scattered multinucleated giant cells or even foci of ossification may be found within nodular fasciitis. Older lesions may lose some of their myxoid character and adopt a somewhat more collagenized appearance. A peculiar histological variant of this lesion, proliferative fasciitis, may appear in children as well as adults; this variant is marked by the presence of large mononuclear cells reminiscent of ganglion cells [9,10].
Clinical Features.
This lesion typically presents as a subcutaneous mass. The overlying skin is movable. Nodular fasciitis is most often painless, although a minority of these lesions may be tender. Frequent sites of involvement include the head and neck region, arms or chest wall, but almost any site may give rise to this reactive process. Nodular fasciitis is, paradoxically, a more rapidly growing lesion than other potentially more aggressive processes. One distinctive paediatric form of nodular fasciitis is known as cranial fasciitis; this condition, largely restricted to infants in the first year of life, is distinguished by the presence of a rapidly growing scalp mass that may erode the outer table of the skull. Cranial fasciitis has the light microscopic attributes of nodular fasciitis and behaves as an altogether benign lesion despite its sometimes rapid growth. Some observers believe that cranial fasciitis may be related to birth trauma and so relegate it to the category of a reactive process [7,11].
Prognosis.
Nodular fasciitis is an entirely benign lesion. These lesions may regress spontaneously, and so recurrence after surgical excision (even an incomplete incisional biopsy) is uncommon.
Differential Diagnosis.
The spindle cells of nodular fasciitis lack either the cytoplasmic eosinophilia typical of a myofibroblastoma or the well-developed storiform architecture of a cutaneous fibrous histiocytoma. While the mitotic activity of nodular fasciitis may at first be disconcerting, it should be borne in mind that nodular fasciitis lacks the cytological atypia that is the hallmark of most true sarcomas. Other considerations for such a clinical presentation include fibromatosis or ganglioneuroblastoma.
Although nodular fasciitis will, in the occasional patient, recur after surgical excision, reports of a recurrent lesion should be greeted with some scepticism and the initial diagnostic material should be reviewed to exclude the possibility that a low-grade sarcoma has been misdiagnosed as nodular fasciitis.
Treatment.
As is the case with myofibromas, a diagnostic biopsy is typically all the therapy that is required for the effective treatment of nodular fasciitis. However, excision of lesions is often sought by affected patients or parents.
References
1 Stout AP. Pseudosarcomatous fasciitis in children. Cancer 1961;14:1216–22.
2 Price EB, Silliphant WM, Shuman R. Nodular fasciitis: a clinicopathologic analysis of 65 cases. Am J Clin Pathol 1961;35:122–36.
3 Montgomery EA, Meis JM. Nodular fasciitis: its morphologic spectrum and immunohistochemical profile. Am J Surg Pathol 1991;15:942–8.
4 DiNardo LJ, Weimore RF, Potsic WP. Nodular fasciitis of the head and neck in children: a deceptive lesion. Arch Otolaryngol Head Neck Surg 1991;117:1001–2.
5 Bernstein KE, Lattes R. Nodular (pseudosarcomatous) fasciitis, a non-recurrent lesion: clinicopathologic study of 134 cases. Cancer 1982;49:1668–78.
6 Shimizu S, Hashimoto H, Enjoji M. Nodular fascilitis: an analysis of 250 patients. Pathology 1984;16:161–6.
7 Lauer DH, Enzinger FM. Cranial fasciitis of childhood. Cancer 1980;45:401–6.
8 Allen PW. Nodular fasciitis. Pathology 1972;4:9–26.
9 Chung ES, Enzinger FM. Proliferative fasciitis. Cancer 1975;36:1450–8.
10 Meis JM, Enzinger FM. Proliferative fasciitis and myositis of childhood. Am J Surg Pathol 1992;16:364–72.
11 Sarangarajan R, Dehner LP. Cranial and extracranial fasciitis of childhood: a clinicopathologic and immunohistochemical study. Hum Pathol 1999;30:87–92.
Infantile (Desmoid-Type) Fibromatosis
Definition.
The locally recurring fibromatoses have traditionally been divided into two groups: those more superficially located lesions (considered below, under plantar–palmar fibromatoses) and the deeply seated lesions (e.g. infantile fibromatosis). Infantile fibromatosis (synonym: deep fibromatosis) is a locally recurring lesion that, although not malignant, may be locally quite aggressive and infiltrate key regional structures including nerves and joints [1–3].
Aetiology.
The aetiology of infantile desmoid-type fibromatosis is unclear, although some familial tendency has been noted in a minority of patients with desmoid-type fibromatoses, a clear pattern of inheritance has not been elucidated for the majority of these patients. Hormonal effects appear to have a role in the growth of these lesions but it is not clear what part these influences play in their genesis.
Pathology.
Infantile desmoid-type fibromatosis comprises a rather homogeneous proliferation of bundles of spindle cells set in a collagenized stroma. Although widely scattered mitotic figures may be found, they are not a prominent feature of infantile fibromatosis. The individual nuclei are often characterized as ‘bland’, a reference to their lack of cytological atypia. In newborn infants, the cellularity of infantile fibromatosis is somewhat greater than that seen in older children or adults.
Clinical Features.
Infantile desmoid-type fibromatosis manifests as firm masses at any age from birth onwards. The lesions are usually painless. Owing to their rapid growth, patients will often promptly seek clinical evaluation. Untreated, the infantile desmoid-type fibromatosis may grow to great proportions. In contrast with the true fibrosarcomas, however, infantile fibromatosis lacks the capacity for metastasis [1–6]. Although infantile desmoid-type fibromatosis may extend towards the superficial regions that fall within the province of the dermatologist, the epicentre of this lesion is rather deeply seated, associated with the muscle or fascia [4–6]. Among the favoured areas for the development of infantile fibromatosis are the shoulder girdle, head, neck and proximal lower extremity. Desmoid fibromatosis will also, in a minority of patients, represent an element of the presentation of Gardner syndrome (familial adenomatous polyposis) with autosomal dominant inheritance, which warrants genetic consultation and DNA analysis of the APC gene if the family history is positive for intestinal tumours [7,8].
Prognosis.
Although infantile desmoid-type fibromatosis does not metastasize, it does show a stubborn tendency to persist and recur locally. In this regard, infantile fibromatosis is the most aggressive of these benign fibrous and myofibroblastic disorders considered here. In addition, infantile desmoid-type fibromatosis may entrap vital structures (including vascular, neural and articular structures) and so interfere with both the quality of life and the lifespan of the affected child.
Differential Diagnosis.
The most difficult disorder to distinguish from infantile desmoid-type fibromatosis is a true (high grade) fibrosarcoma. Although the cellularity of a fibromatosis (developing in an older child) falls short of that of the usual (congenital) infantile fibrosarcoma, newborn infants with infantile desmoid-type fibromatosis may show a more cellular pattern than that seen in older patients, resembling a congenital fibrosarcoma. This distinction may be extremely difficult with only limited material for diagnosis.
One other critical lesion that must be considered in the differential diagnosis is that of the low grade fibrosarcoma: a lesion that has been poorly characterized to date. The low grade fibrosarcomas behave similarly to fibromatoses in many regards, but with one addition: the low grade fibrosarcomas possess a small, but real, likelihood of metastasizing.
Myofibromas, neural tumours and reactive fibroblastic proliferations may also be considered in the differential diagnosis of infantile fibromatosis.
Treatment.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


