BP, bullous pemphigoid; PF, pemphigus foliaceus; PV, pemphigus vulgaris.
History.
At the end of the 18th century, the name pemphigus was used for the first time. Until the early 20th century, all chronic blistering diseases were designated as pemphigus [1]. The disease pemphigus was characterized histologically by Civatte [2]. In 1964, the characteristic immunopathology was described, which provides the opportunity to make a more definite diagnosis by performing immunofluorescence examination [3].
In 1955, the first well-defined case report of pemphigus vulgaris in a child was published [4]. The earliest paediatric case report of pemphigus vulgaris, with immunofluorescence findings, appeared in 1969 [5] and of pemphigus foliaceus in 1968 [6]. In the world literature, more than 55 cases of pemphigus vulgaris in children, the majority of whom were over 10 years of age, have been reported [7]. No male or female predilection has been observed.
The non-endemic variant of pemphigus foliaceus in children is even more rare than pemphigus vulgaris. Around 30 cases have been reported. Most of them were children under 10 years of age [8].
Aetiology and Pathogenesis.
Pemphigus is a disease of intercellular adhesion of epidermal cells caused by pathogenic circulating autoantibodies [9]. Epidermal cells adhere and communicate by four major groups of junctions: desmosomes, adherens junctions, tight junctions and gap junctions [9]. The desmosomes consist of transmembrane components, the desmosomal cadherins (desmogleins and desmocollins), which bind to the attachment plaque containing the armadillo proteins (plakophilin and plakoglobin), and the plakins (desmoplakin, envoplakin, periplakin and plectin) [10] (Fig. 91.1). In the epidermis, desmosomes are associated with keratin intermediate filaments. The second form of junctions, the adherens junction, contains the so-called ‘classic cadherins’ (E-cadherins) and the plaque proteins (plakoglobin and β-catenin) among other proteins [11]. Adherens junctions are associated with actin microfilaments. The pemphigus antigens have been identified as the cadherin antigen called desmoglein-3, a 130 kDa glycoprotein, and desmoglein-1, a 160 kDa glycoprotein [12]. The antibodies in mucocutaneous pemphigus vulgaris patients react with these antigens. The precise mechanism for acantholysis when pemphigus IgG binds to the cell surface is as yet unknown. Ultrastructurally, widening between epidermal cells appears first between desmosomes (spongiosis) [13] and later also hypoplastic desmosomes split in half [14].
Fig. 91.1 Diagram of desmosome and adherens junctions of keratinocytes with pemphigus autoantigens in yellow.
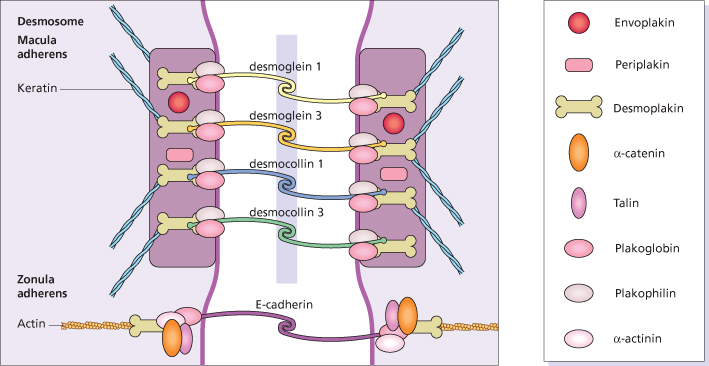
In pemphigus foliaceus the antigen is desmoglein-1 [9]. The antibodies from patients with pemphigus vulgaris and pemphigus foliaceus bind selectively to desmosomes and adherens junctions of the epidermis. The binding of antibodies to desmoglein-3 in pemphigus vulgaris is in the lower layer of mucous membrane epithelium not compensated by enough desmoglein-1, whereas the binding of desmoglein-1 in pemphigus foliaceus is in the upper epidermis not compensated by enough desmoglein-3 [15]. This desmoglein compensation hypothesis explains the following histological picture: the blister of pemphigus vulgaris occurs in the suprabasal layer, and the blister in pemphigus foliaceus is in the stratum granulosum or even in the subcorneal layer. The desmoglein compensation hypothesis also explains why pemphigus vulgaris patients with antibodies against desmoglein-3 only have their mucous membranes involved, while those with additional antibodies against desmoglein-1 also have skin lesions.
Pemphigus Vulgaris
Pathology.
The characteristic histopathological picture of pemphigus vulgaris is best found in a fresh lesion and shows an intraepidermal separation of basal cells from spinous cells. The basal cells remain attached to the basement membrane but have laterally lost contact, giving the basal layer a characteristic ‘tombstone’ appearance. The suprabasal bulla contains acantholytic keratinocytes. In the superficial dermis, a sparse perivascular infiltrate of mixed inflammatory cells is present. Acantholysis and suprabasal clefts may extend into the follicular units. Epidermal spongiosis with many eosinophils (eosinophilic spongiosis) may be the first sign of pemphigus vulgaris, later evolving into blisters filled with eosinophils and acantholytic cells.
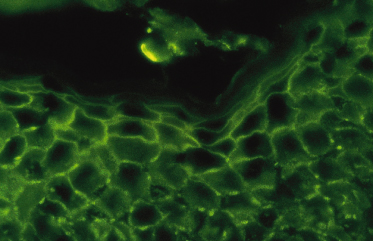
The antibodies may be detected in the epidermis by direct immunofluorescence of a biopsy from perilesional or uninvolved skin. Direct immunofluorescence shows depositions of IgG, IgA and sometimes C3 in an intercellular pattern throughout the epidermis (Fig. 91.2) [16]. It has been shown that the pemphigus-specific immunofluorescence pattern is also demonstrable in the outer root sheaths of plucked anagen hairs [17]. This seems to be an easy way to obtain direct immunofluorescence data in a child. Circulating IgG and IgA antibodies, which bind to desmogleins, are detectable by ELISA [16]. Antidesmoglein-1 antibody ELISA values are more closely correlated than antidesmoglein-3 antibody ELISA values with the course of the disease in patients with pemphigus vulgaris or pemphigus foliaceus [18].
Fig. 91.2 Positive direct immunofluorescence of the skin with anti-IgG staining in a child with pemphigus vulgaris.
Clinical Features.
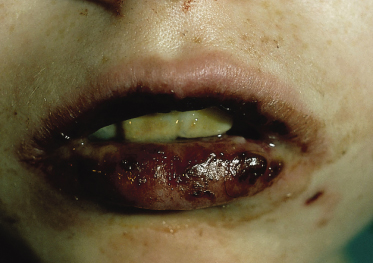
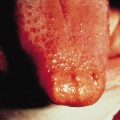
Stomatitis is the presenting sign in more than 50% of the children with pemphigus vulgaris (Fig. 91.3). The lesions of the oral mucosa vary from superficial blisters to painful ulcers, accompanied by dysphagia. Subsequently, bullae on the skin may develop. In some cases, however, oral lesions are the only manifestation. In 80% of children with pemphigus vulgaris, the mucosa, as well as the skin, is affected in the course of the disease. In some children, skin blisters may be the sole symptom of pemphigus vulgaris and no mucous membrane lesions may be present. The skin blisters, on normal appearing skin or on an erythematous base, are typically flaccid and filled with a clear or slightly haemorrhagic fluid. The Nikolsky sign, meaning that a blister can be provoked by tangential pressure on the normal-appearing skin adjacent to a bulla, is often positive; an existing blister can also be spread by pressure (Asboe-Hansen sign). The blisters may occur over the whole integument and, unless secondary impetiginization occurs, they heal without scarring.
Fig. 91.3 Erosive stomatitis as a presenting sign in pemphigus vulgaris.
Reproduced with permission from the Annales de Dermatologie et Vénéreologie.
There are isolated case reports of drug-induced pemphigus vulgaris in children. In these patients, the disease has been associated with isotretinoin [19], enalapril [20] and vaccination (diphtheria tetanus cocktail) [21]. Montelukast and carbamazepine have also been linked to the induction of pemphigus vulgaris in individual paediatric cases [22,23].
One child with a combination of pemphigus vulgaris and Graves disease has been reported. As withdrawal of propylthiouracil and levothyroxine did not ameliorate the pemphigus, drug-induced pemphigus is not probable in this case [24].
Prognosis.
Before the introduction of systemic corticosteroid therapy, pemphigus vulgaris was considered a fatal disease [25]. In adults, early diagnosis and institution of treatment have been shown to improve the outcome. The mean diagnostic and therapeutic delay in published case reports of children with pemphigus vulgaris is approximately 6 months [26]. In children, no relation between prognosis and the delay before diagnosis has been demonstrated. The prognosis is variable. The majority of children described suffered from a severe form of the disease, requiring high doses of corticosteroids. In 80% of the reported cases, it was necessary to continue with a maintenance therapy to prevent recurrences. Of the reported cases, 20% are in remission without any treatment during follow-up periods of 15 months to 19 years [27]. Only three reported children died, two from sepsis and one from respiratory insufficiency [26]. In a 15-year-old girl, pemphigus vulgaris was followed by systemic lupus erythematosus 5 years later [28]. Although pemphigus vulgaris is still a very serious disease in children, the prognosis is relatively good, especially when compared with that in the pre-corticosteroid era.
Differential Diagnosis.
For mucosal lesions, the differential diagnosis includes herpes gingivostomatitis, aphthous stomatitis, erythema multiforme, lichen planus, pyostomatitis vegetans (as part of ulcerative colitis and Crohn disease), Behçet disease, lupus erythematosus and mucous membrane pemphigoid [7]. For the skin lesions, the initial differential diagnosis includes bullous impetigo, erythema multiforme, toxic epidermal necrolysis, bullous pemphigoid, dermatitis herpetiformis, drug eruption and bullous contact dermatitis [5]. A definite diagnosis can be made with a combination of histopathology and immunofluorescence examination. An infectious origin for mucosal as well as skin lesions must be excluded by viral and bacterial cultures.
Treatment.
In mild forms of pemphigus vulgaris, topical treatment with corticosteroid ointment (triamcinolone acetonide or more potent corticosteroids) twice daily may be sufficient. In isolated lesions, intralesional corticosteroids can also be used. In the majority of cases, however, systemic treatment with corticosteroids is required. The dosage of prednisolone in milligrams per kilogram of bodyweight per day should be guided by the degree of cutaneous and mucous membrane involvement, as well as, to some extent, by the antibody titre measured by indirect immunofluorescence. The main goal is to obtain control of the disease with the lowest possible dosage of prednisolone. Prednisolone equivalents of 0.5–2 mg/kg bodyweight per day may be needed initially to suppress disease activity; 1–4 weeks after the disease is under control, the prednisolone dosage should be tapered and, if possible, an alternate-day schedule should be followed. This is done stepwise, titrating the dose to the clinical response. In patients with very extensive cutaneous lesions (25–70% body area), intravenous steroid as pulse treatment has been advocated [29]. This pulse treatment consists of 0.5–1 g of methylprednisolone in 5% dextrose, given intravenously over 1–2 h on three consecutive days. These pulses are repeated once per month. In between, 10–20 mg of prednisolone may be given daily. In adults pulsed dexamethasone therapy was found to have no benefit above placebo as adjuvans to prednisolone and azathiorptine [29].
Steroid-sparing options include azathioprine and cyclophosphamide. These immunosuppressive drugs become effective after 4–8 weeks. Toxic effects on bone marrow and liver may occur. Azathioprine is the most commonly used adjuvant in children [30], and no serious side-effects have been reported. However, acute bone marrow depression may occur in children with genetically determined thiopurine methyltransferase (TPMT) deficiency. If possible, TPMT activity should be determined and dosing of azathioprine adapted to the level of activity [31]. Cyclophosphamide administration is an alternative, but it may lead to infertility in both males and females. Moreover, this drug may cause haemorrhagic cystitis. There is an association between the use of cyclophosphamide and the occurrence of bladder malignancy, in particular when cumulative dosages of more than 20 g have been given [32]. Methotrexate treatment has been largely abandoned because of the high dosage required, the high rates of severe infections and the impairment of cutaneous healing [33]. Although ciclosporin has been successfully used in individual cases of adults and children with pemphigus vulgaris [34], there is insufficient evidence that it is of any help in pemphigus in general [35]. In one case report, the occurrence of keloids was suggested to be caused by the use of ciclosporin [36].
The use of gold has been reported to be successful in children [37]. In a rare case associated with antidesmoglein and antidesmoplakin antibodies without underlying malignancy, gold proved ineffective [38]. Gold treatments may be associated with a relatively high risk of adverse reactions. As rheumatologists generally have more experience with gold treatment, a multidisciplinary approach may be useful [37].
Dapsone is safer than most adjuvant agents used in the treatment of pemphigus and it has been given in children with pemphigus vulgaris [39], and its efficacy in adult pemphigus patients has recently been evaluated to be reasonably good although statistically not reaching significance [40]. Glucose-6-phosphate dehydrogenase deficiency should be excluded before its use.
Mycophenolate mofetil is a reversible non-competitive inhibitor of the key enzyme in purine biosynthesis, which suppresses the proliferation of T- and B-lymphocytes. It has been used rarely in children for pemphigus [41,42]. The potential adverse effects are mainly gastrointestinal and those due to immunosuppression (myelosuppressive).
Various more direct ways to decrease the antibody titres in pemphigus have been described. Plasmapheresis and immunoapheresis offer a considerable steroid-sparing effect and, as adjuvants, they have been used in children with variable results [42,43]. High-dose intravenous immunoglobulin (IVIG) therapy has been demonstrated in placebo-controlled trials in adult pemphigus patients to control active disease [44]. Its use in children is not yet described. The use of IVIG in children in general is proven to be well tolerated, in particular when premedication with hydrocortisone and an antihistamine is given [45,46]. The indication is for unresponsiveness of pemphigus to conventional immunosuppressive treatments or significant side-effects from these therapies. Rituximab, a monoclonal antibody against CD20 present on B-cells, appears to be a very effective drug against pemphigus [47] and it also has been used succesfully in juvenile pemphigus [42,48,49].
In conclusion, the advantages of immunosuppressives should be weighed against the disadvantages in the individual case. If the patient is in remission, prednisolone is usually tapered more quickly than adjuvant therapy [34]. Lowering of antibody levels by plasmapheresis, immunoapheresis, high-dose intravenous immunoglobulins or rituximab are alternatives when more traditional treatments are ineffective or show serious side-effects.
Pemphigus Vegetans
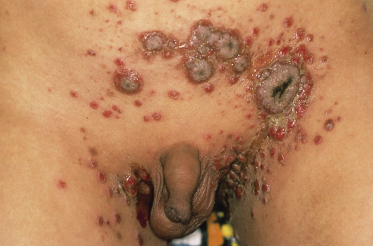
Pemphigus vegetans is a rare variant of pemphigus vulgaris characterized by verrucous, intertriginous plaques during the healing phase. The vegetating lesions seem to represent a special reactive pattern of the skin. Specific areas, such as the groin and the axillary regions, are more prone to this vegetating tendency. Histologically, pemphigus vegetans lesions show acantholysis, in combination with papillomatosis of the dermal papillae and a hyperplastic epidermis. Immunofluorescence of perilesional skin is indistinguishable from that in pemphigus vulgaris. Clinically, lesions of pemphigus vulgaris in flexural sites or lesions resistant to treatment may develop into pemphigus vegetans. In these places, painful vegetating plaques develop (Fig. 91.4). The involved areas are sometimes studded with pustules, which produce serum and crusts and may become secondarily infected. Lesions may become malodorous and often heal with hyperpigmentation. The prognosis is probably similar to that of pemphigus vulgaris. Only some cases have been reported in children [50–53]. Treatment is identical to that used in pemphigus vulgaris.
Fig. 91.4 Vegetating plaques in a boy with pemphigus vegetans.
Reproduced with permission from the British Journal of Dermatology.
In the differential diagnosis, pyoderma vegetans, as well as benign and malignant acanthosis nigricans, must be considered. They can be excluded by histology and immunofluorescence.
Pemphigus Foliaceus
Definition.
Pemphigus foliaceus is a superficial variant of pemphigus in which autoantibodies against desmoglein-1 induce superficial blistering [54].
Pathology.
Histopathological examination of a biopsy from an intact blister (often difficult to find in superficial forms of pemphigus) shows an intraepidermal blister at the level of the stratum granulosum, or even a subcorneal blister. In the blister, acantholytic cells and varying numbers of neutrophils are seen. In the dermis, a slight to moderate inflammatory infiltrate with a predominance of eosinophils is usually present. Histological examination can be misleading unless an early, uninfected lesion is biopsied.
Immunofluorescence examination of perilesional or uninvolved skin is essentially the same as that of pemphigus vulgaris, showing depositions of IgG, and sometimes IgA and C3, in an intercellular pattern throughout the epidermis. By indirect immunofluorescence, circulating IgG antibodies, which bind to the surface of epithelial cells, are detected in most affected children.
Clinical Features.
Initially, in one-half of the cases, erythema of the scalp with scaling occurs, which may develop into oozing, crusts and blisters. The lesions may cause pain and discomfort. Over a period of weeks the eruption may progress to involve the trunk and upper limbs. In other patients, the earliest lesions develop on the trunk or the extremities. In contrast to pemphigus vulgaris, the mucous membranes are only very rarely involved. Photodistribution has been described [55]. Crusted plaques and erosions predominate; bullous or vesicular lesions are rarely found, due to their fragility (Fig. 91.5). The Nikolsky and Asboe-Hansen signs (see above) are positive. The crusted lesions may follow an arcuate pattern. Healing occurs without scarring. Rarely, an exfoliative erythrodermic variant may occur and this form follows a rather aggressive course [56].
Induction of pemphigus foliaceus by amoxicillin was reported in two paediatric cases [57,58]. In these children, the skin lesions started after 8 and 10 days, respectively, of amoxicillin treatment and in both children an exacerbation of the disease was induced by a subsequent treatment with this antibiotic.
In one case, a co-existence of pemphigus foliaceus and cytomegalovirus infection was described. In this haemophilic child, the cytomegalovirus antibody titres reflected the activity of the pemphigus [59]. An association between pemphigus foliaceus and untreated Graves disease has been reported in one patient [60]. One 6-year-old boy developed pemphigus foliaceus after mismatched cord blood transplantation in the treatment of Philadelphia chromosome-positive B-cell leukaemia [61].
Prognosis.
Long follow-up periods of children with pemphigus foliaceus have not been reported (range 2 months to 4 years [55]); one reported case died from the skin disease [62]. Most children required continuous treatment during follow-up. In very few children, topical treatment with corticosteroids was sufficient to suppress disease activity. In 25% of the children, the topical or systemic treatment was stopped without recurrence of the disease. Based on these observations from the literature, the prognosis of pemphigus foliaceus seems to be roughly similar to that of pemphigus vulgaris.
Differential Diagnosis.
In the early stage of the disease, pemphigus foliaceus is often confused with impetigo (excluded by a bacterial culture) or, if the lesions are located on the scalp, with seborrhoeic dermatitis or psoriasis. In generalized eruptions, erythema multiforme, toxic epidermal necrolysis, pustular psoriasis, drug eruption and bullous forms of contact dermatitis may be considered. The combination of histopathology and immunofluorescence is necessary to make the correct diagnosis.
Treatment.
In rare cases, it may be possible to suppress the disease with topical corticosteroids alone, such as triamcinolone acetonide or stronger [57,63,64]. In more extensively involved patients, systemic treatment with corticosteroids is required. Daily doses of 1–2 mg/kg are sufficient, but according to some case reports up to 5 mg/kg of prednisolone may be necessary to prevent the occurrence of new lesions and to induce the drying of existing ones. The additive or steroid-sparing effect of other immunosuppressive drugs such as azathioprine and cyclophosphamide may be helpful, but their advantages have to be weighed against possible side-effects, especially in the long term. Dapsone was used in five children [58,61,65–68] and proved to be ineffective. The efficacy of sulphapyridine is difficult to establish as it has been reported only once and it was given in combination with azathioprine and prednisolone [69]. Sublesional triamcinolone injection was reported to be effective in one patient [70]. A CD-20 antibody (rituximab) in a dosage of 375 mg/m2/wk was successfully given in two seriously involved cases [56,61]. Other treatment options such as high-dose immunoglobulins intravenously and mycophenolate mofetil appeared to be not effective enough even when combined with corticosteroids [56,61], although some recent data indicate that mycophenolate mofetil can be as effective as azathioprine [71].
Brazilian Pemphigus (Fogo Selvagem)
Definition and History.
Brazilian pemphigus (fogo selvagem), also known as endemic pemphigus, is a form of pemphigus foliaceus which was identified at the end of the 19th century [72] and is confined to certain regions of Brazil, Venezuela, Colombia and Tunisia. Clinically and immunologically, it is identical to the idiopathic form of pemphigus foliaceus. The only difference is in its epidemiology [73].
Aetiology and Pathogenesis.
The production of autoantibodies against desmoglein-1 (the same antigen as that in pemphigus foliaceus) is an early step in the pathogenesis of fogo selvagem. A high percentage of normal subjects in an endemic region have a humoral immune response to desmoglein-1 [74], whereas 36% also develop non-pathogenic antibodies against desmoglein-3 [75]. Genetic factors, such as human leucocyte antigen (HLA) alleles in combination with epitope spreading, may result in expression of the disease [74]. The endemic character of pemphigus foliaceus in certain regions is probably caused by an environmental factor. The black fly is considered an insect vector and plays an important role in the origin of the disease [76]. Whether the saliva of the insect contains antigens that cross-react with desmoglein-1 or whether the fly carries an infectious agent that initiates fogo selvagem is not yet known [77]. Specific HLA alleles make people susceptible to pemphigus foliaceus; HLA-DRl alleles appear to be the susceptibility gene, which, in combination with absence of the protective gene HLA-DQw2, predisposes the subject to the disease [78].
Pathology.
The histopathological and immunofluorescence findings in endemic pemphigus foliaceus are indistinguishable from those in sporadic pemphigus foliaceus.
Clinical Features.
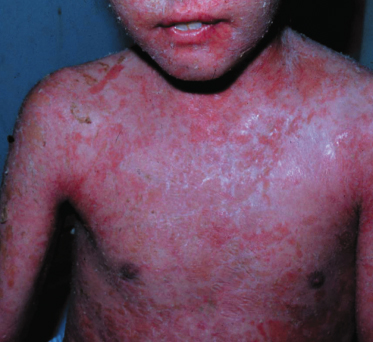
Sporadic and endemic pemphigus foliaceus have a similar clinical appearance, although endemic pemphigus is more common in young people: 30% of the cases occur in individuals of less than 20 years of age [79]. The typical patient with the endemic form of pemphigus foliaceus is a farmer or family member of a farmer of young age (usually a child or young adult). He or she lives near rivers or creeks and spends a lot of time outdoors. There is a high frequency of familial cases. These data do not imply that the disease is contagious, because other close contacts such as hospital workers are not affected [72]. The primary lesion, a superficial vesicle, ruptures easily, leaving superficially denuded areas (Figs 91.6 and 91.7). Lesions always occur on the head and/or neck area and then spread acrally. Mucosal lesions are rare. The distribution strongly resembles that of sun exposure. Evolution is often quite slow, with skin lesions developing over several weeks or months; the disease is only rarely acute and fulminant [73]. Chronic cases or cases with localized disease often show verrucous or vegetating plaques. Hyperpigmentation syndromes are seen in patients undergoing remission. This hyperpigmentation may be restricted to areas of previous lesions or may be diffuse and involve previously unaffected skin [73].
Fig. 91.6 An 11-year-old white boy with fogo selvagem of the bullous exfoliative type.
Courtesy of Valeria Aoki, Assistant Professor in Dermatology, Department of Dermatology, University of São Paulo, Brazil.
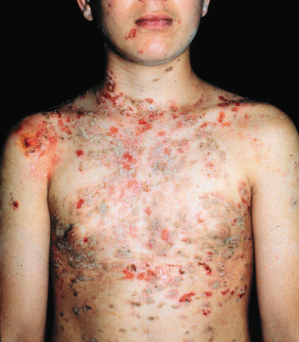
Fig. 91.7 A 6-year-old white girl showing an erythrodermic type of fogo selvagem.
Courtesy of Valeria Aoki, Assistant Professor in Dermatology, Department of Dermatology, University of São Paulo, Brazil.
In children affected by endemic pemphigus foliaceus, normal growth becomes arrested, and if the disease persists for long periods they may suffer from dwarfism. If children are treated with prednisolone, they show catch-up growth and achieve near normal stature [72].
Prognosis.
In the 1930s and 1940s, before steroids were available, approximately 50% of adult patients died of the disease [79], whereas about 20% underwent spontaneous remission irrespective of the extent of the disease. At present, less than 10% of the patients die, and those who do may succumb to complications of steroid therapy [72]. Unfortunately, no specific prognostic data are available concerning children. Male patients who have had the disease as a child may show azoospermia [72].
Differential Diagnosis and Treatment.
The differential diagnosis is the same as for sporadic pemphigus foliaceus. As in the other forms of pemphigus, the mainstay of treatment is systemic administration of prednisolone. In resistant cases, or in local vegetating and verrucous plaques, adjuvant topical treatment with fluorinated corticosteroids promotes healing. Clinical experience in South America suggests that systemic triamcinolone is superior to prednisolone, with a lower incidence of side-effects [72]. Immunosuppressive agents and gold compounds have been used successfully, but bear the above-mentioned risks.
Pemphigus Erythematosus
Pemphigus erythematosus is a pemphigus foliaceus variant with IgG and C3 at the basement membrane zone, in addition to IgG in an intercellular epidermal pattern. Circulating antibodies against intercellular substance, and also antinuclear antibodies, may be detected. Only two well-analysed childhood cases have been described [80,81]. The disease may start as a malar eruption reminiscent of a lupus erythematosus-like butterfly rash. Blisters may be present and spread to the rest of the body.
Pemphigus Neonatorum
Definition.
Pemphigus neonatorum is pemphigus as a neonatal disease due to the transplacental passage of autoantibodies against intercellular substance from the mother. It must be distinguished from primary pemphigus vulgaris in a neonate [82].
History.
The first well-documented report with immunofluorescence of a child with pemphigus born from a mother with pemphigus vulgaris was published in 1975 [83]. As direct immunofluorescence on neonatal skin was positive for intercellular IgG antibodies, it appeared to be a confirmation of experiments in which IgG antibodies from patients with pemphigus were given to mice, causing a bullous eruption that was indistinguishable from pemphigus [84]. To date, more than 20 cases of neonatal pemphigus vulgaris have been published and several cases of neonates affected with pemphigus foliaceus [85].
Aetiology and Pathogenesis.
The study of infants with histologically and immunologically proven pemphigus lesions, born from mothers with circulating antibodies to intercellular substance, provides further evidence for the pathogenic role of the IgG antibodies against intercellular substance in the acantholysis seen in pemphigus. The rarity of pemphigus foliaceus in neonates born from mothers with pemphigus foliaceus may be explained by the fact that, in neonates, desmoglein-3 is present throughout the epidermis [86]. In contrast, desmoglein-3 is only present in basal and immediate suprabasal layers of adult skin. The anti-desmoglein-1 antibodies in children born from pemphigus foliaceus mothers interfere with the function of desmoglein-1, but in these children the desmoglein-3 present throughout the epidermis may largely compensate for this loss of function.
Pathology.
The histopathological picture of a skin lesion in the neonate from a mother with pemphigus vulgaris is that of pemphigus vulgaris. Direct immunofluorescence shows intercellular deposition of IgG and sometimes C3 in the skin. Indirect immunofluorescence can be positive, but the antibody titre is equal to or lower than that in the mother. In neonatal pemphigus foliaceus, the histopathological picture is that of pemphigus foliaceus in adults.
Clinical Features.
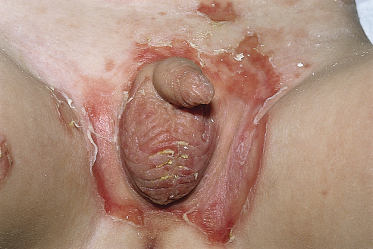
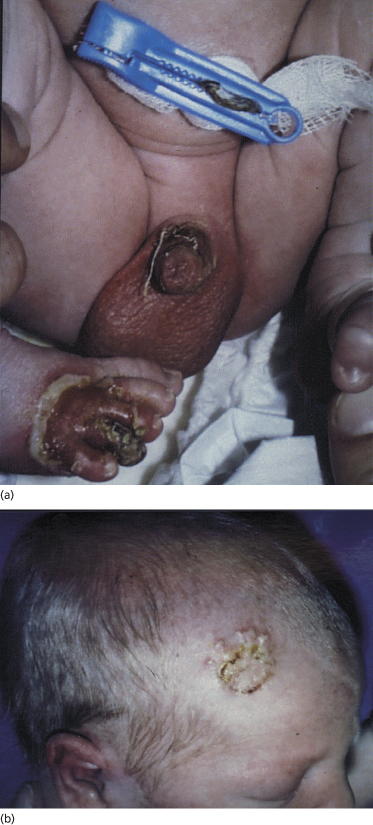
Most neonates with pemphigus vulgaris present at birth with bullae, vesicles and erosions of the skin (Fig. 91.8). Mucous membrane involvement was reported in some infants. Four case reports concern stillborn children, but the cause of intrauterine death in these children has not been established [87–90]. There seems to be no connection between stillbirth and the extent of the skin surface involved or antibody titre. Mothers do not always have active disease when the affected child is born [83,91]. In neonatal pemphigus foliaceus, the clinical picture is similar to that in neonatal pemphigus vulgaris, but oral lesions do not occur.
Fig. 91.8 Erosions on the penis and right foot (a) and vesicles and crusts on the head (b) in a 2-day-old child with neonatal pemphigus.
Courtesy of T. Parlovsky and reproduced with permission from the Journal of the American Academy of Dermatology.
Prognosis.
When the child is born alive, the prognosis in both pemphigus vulgaris and pemphigus foliaceus of the newborn seems good [92]. Healing of the skin lesions occurs within 3 weeks, and is most probably related to the decrease in maternal antibodies. With resolution of active lesions, residual hyperpigmentation may remain. In four case reports of pemphigus vulgaris neonatorum, stillbirth is described. However, mothers with pemphigus vulgaris may also give birth to normal children [93–95].
Differential Diagnosis.
If a diagnosis of pemphigus vulgaris or pemphigus foliaceus is established in the mother, pemphigus must be considered in an infant with blisters. Confirmation by histology and immunofluorescence (direct and indirect) is necessary. Staphylococcal scalded skin syndrome, neonatal herpes simplex infection, congenital bullous diseases such as epidermolysis bullosa and bullous mastocytosis should be included in the differential diagnosis. In a child born from a healthy mother, primary pemphigus vulgaris in a neonate is possible [96].
Treatment.
As spontaneous recovery is reported in all cases within 3 weeks, no treatment is necessary. Topical antibacterial therapy may be necessary if secondary infection of the lesions occurs.
Paraneoplastic Pemphigus
Paraneoplastic pemphigus, also known as paraneoplastic autoimmune multiorgan syndrome, is an autoimmune mucocutaneous disease occurring in association with neoplasia which was identified in 1990 by Anhalt [97]; the clinical, histological and immunological features of pemphigus are present. The detection of serum antiplakin antibodies is the most reliable diagnostic tool.
At least 15 paediatric cases have been published [98–101]. All of these children developed debilitating painful stomatitis, not responsive to treatment with high-dose corticosteroids or other immunosuppressives. In most children, oral mucosa and other mucosal surfaces (e.g. genital mucosa and severe conjunctivitis) are involved. The patient may develop lichenoid lesions or erythematous, crusted or erosive lesions, sometimes involving large body areas. Skin lesions may either precede [99] or start after the development of malignancy [98]. In the majority of children, paraneoplastic pemphigus is associated with Castleman disease [100] but association with T-cell lymphoblastic lymphoma and sarcoma has also been described. In most cases, severe respiratory involvement (bronchiolitis obliterans) develops; this feature is a major factor in the high mortality rate of the disease. Children who did survive did not have lung involvement or had successful lung transplantation [100,101].
Erythema multiforme and acute febrile ulceronecrotic Mucha–Haberman disease may resemble paraneoplastic pemphigus in many respects. Differentiation is based on histological and immunofluorescent characteristics.
The treatment of the neoplasia (e.g. T-cell lymphoblastic lymphoma [98], Castleman tumour [99]) and lung disease is essential for survival. The therapy for skin and mucosa is the same as that in pemphigus vulgaris.
Immunoglobulin A Pemphigus
Immunoglobulin A (IgA) pemphigus is a rare type of pemphigus characterized by a vesiculobullous eruption. It is classified, based on histological and immunological characteristics (immunofluorescence and immunoblotting studies), into two major subtypes: subcorneal pustular dermatosis (SPD) and intraepidermal neutrophilic IgA dermatosis (IEN). In the SPD type, IgA autoantibodies react with desmocollin in the upper levels of the epidermis. In the IEN type the target antigen has not been identified [102]. Several paediatric cases have been described, the youngest being 1 month old when the disease started [102–109]. In one child, the antibodies were directed against desmoglein-1, the antigen of pemphigus foliaceus [105].
Clinically, the eruption consists of superficial small blisters on an erythematous background. As the bullae are very fragile and evanescent, they are often replaced by crusts and erosions. The lesions may coalesce to form ring-like or serpiginous patterns, sometimes with blisters at the periphery. The mucous membranes are mostly spared. One unique case with skin and mucous membrane involvement has been described; this patient developed IgA and IgG antibodies against desmoplakin-I/II, desmocollin and desmoglein-3 [109].
The prognosis in children seems to be good, although most children still had active disease at the end of follow-up (5 years or less) [102–109].
The main differential diagnostic considerations – impetigo bullosa, linear IgA dermatosis, subcorneal pustular dermatosis, pustular psoriasis and pemphigus foliaceus – are mainly ruled out by bacterial culture or immunofluorescence [103]. The best mode of treatment seems to be dapsone (50–100 mg per day) which may be used, if necessary, in combination with low-dose prednisolone (0.5 mg/kg per day or lower) or topical corticosteroids [103]. Etretinate (1 mg/kg per day) has also been reported to be effective [104]. The medication should be gradually tapered. Pristinamycine, a macrolide antibiotic, was also shown to be effective in one paediatric case [106].
Hailey–Hailey Disease (Chronic Benign Familial Pemphigus)
Chronic benign familial pemphigus is a rare autosomal dominantly inherited skin disease (MIM 16960). The first description was given by the brothers Hailey [110] in 1939.
The disease is caused by mutations in the ATP2C1 gene, which plays an important role in the function of the Ca2+ pump [111]. Mutations in ATP2C1 have been established as the cause of the disease which has provided compelling evidence that Ca2+- and Mn2+-ATPases play a key role in the assembly of desmosomes in the epidermis. Defective cell adhesion may be secondary to abnormalities in the recruitment, folding, sorting or trafficking of desmosomal proteins or the stabilization of the junctions through links with the cytoskeleton [112].
Histopathologically, the epidermis of lesional skin shows widespread suprabasal acantholysis leading to intraepidermal vesicles and bullae, while the epidermis shows the appearance of a dilapidated brick wall. Moreover, narrow strands of epidermal cells proliferate downwards and a mild dermal perivascular infiltrate may be present [113]. In contrast to autoimmune pemphigus, direct and indirect immunofluorescence examination is negative [114].
Clinical symptoms of chronic benign familial pemphigus usually present in the second to fourth decades of life [115]; symptoms in the first decade are very rare [116]. Sites of predilection include the neck, groin, perineum and axillary region, all areas where minor trauma may induce skin changes. Lesions may present as plaques with a collarette-like scaly border, crusted erosive lesions with vesicles and pustules, small erythematous scaly plaques and hypertrophic flexural lesions [115]. A blister can be provoked by tangential pressure on normal-appearing skin adjacent to a lesion (Nikolsky sign). The involved areas heal without scarring. Multiple longitudinal white bands in the nails may occur, even before other signs of the disease are present [115,117].
Physical activity is limited by pain and patients may complain of itching. An unpleasant smell may emanate from involved skin. Friction, heat or sweating, and to a lesser degree stress and sun exposure, exacerbate the disease [115].
Bacterial skin infection is a frequent complication, but also herpes virus (even Kaposi varicelliform eruption) [118] and mycotic infections [114] occur. Relapses of variable duration tend to be less intense and less frequent over time. The prognosis is favourable but chronic benign familial pemphigus is an unpredictable illness.
Differential diagnosis includes candidal intertrigo and impetigo, contact dermatitis and Darier disease. In Darier disease, lesions are not induced by minor trauma and the patients often show palmar pits, keratotic palmar papules and keratotic papules on the trunk. The nail changes in Darier disease, in contrast with those in chronic benign familial pemphigus, are symptomatic.
Treatment consists of moderate to potent topical corticosteroids, sometimes in combination with topical antibacterial agents; oral antibiotics may be required. In treatment-resistant cases, dapsone, etretinate, psoralens and ultraviolet A (PUVA), thalidomide, methotrexate and even ciclosporin have been used [119]. Unfortunately, none of these is effective on a long-term basis. Dermabrasion and carbon dioxide laser vaporization give more long-lasting results [119,120] and are less drastic procedures than excision of involved skin. Recently, topical aminoglycoside such as gentamicin, with the objective of inducing readthrough of a pathogenic nonsense mutation, was found to be far more effective in inducing remission in a Hailey–Hailey disease carrying a premature stop mutation than an accepted topical disinfectant [121].
References
1 Thivolet J. Pemphigus: past, present and future. Dermatology 1994;189(Suppl. 2):26–9.
2 Civatte A. Diagnostique histopathologique de la dermatite polymorphe douloureuse ou maladie de Duhring–Brocq. Ann Dermatol Syph 1943;3:1–30.
3 Beutner EH, Jordon RE. Demonstration of skin antibodies in sera of pemphigus vulgaris patients by indirect immunofluorescent staining. Proc Soc Exp Biol Med 1964;117:505–10.
4 Mintzer IJ, Rubin Z. Pemphigus vulgaris in a 13-year-old girl. NY State J Med 1955;55:1903–7.
5 Jordon RE, Ihrig JJ, Perry HO. Childhood pemphigus vulgaris. Arch Dermatol 1969;99:176–9.
6 Kierland RR, Perry HO, Winkelmann RK et al. Pemphigus foliaceus or erythema multiforme. Arch Dermatol 1968;98:316–17.
7 Prendiville JS, Israel DM, Wood WS et al. Oral pemphigus vulgaris associated with inflammatory bowel disease and herpetic gingivostomatitis in an 11-year-old girl. Pediatr Dermatol 1994;11:145–50.
8 Kanwar AJ, Kaur S. Pemphigus in children. Int J Dermatol 1991;30:343–6.
9 Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N Engl J Med 2006;355:1800–10.
10 Green KJ, Simpson CL. Desmosomes: new perspectives on a classic. J Invest Dermatol 2007;127:2499–515.
11 Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol 2007;127:2525–32.
12 Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 1991;67:869–77.
13 Diercks GF, Pas HH, Jonkman MF. The ultrastructure of acantholysis in pemphigus vulgaris. Br J Dermatol 2009;160:460–1.
14 Wang W, Amagai M, Ishiko A. Desmosome splitting is a primary ultrastructural change in the acantholysis of pemphigus. J Dermatol Sci 2009;54:59–61.
15 Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest 1999;103:461–8.
16 Mentink LF, de Jong MC, Kloosterhuis GJ, Zuiderveen J, Jonkman MF, Pas HH. Coexistence of IgA antibodies to desmogleins 1 and 3 in pemphigus vulgaris, pemphigus foliaceus and paraneoplastic pemphigus. Br J Dermatol 2007;156:635–41.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


