Skin lesions
Bone marrow histologya
Peripheral blood counts
Serum tryptase
Present, MC infiltrates, c-kit mutation in lesional skin
Negative, no MC infiltrates
Normala
<20 ng/mLa
Skin lesions
Bone marrow histology
Bone marrow cytology (smears)
CD2/CD25 on bone marrow MC
Peripheral blood counts
Serum tryptase
Liver/spleen/lymph nodes
Present (vast majority)
Multifocal MC infiltrates
Bone marrow MC <20%, low grade
Found
Normal or slightly abnormal
>20 ng/mL
Organomegaly may be found
WHO/FAB criteriab
AHNMD
Skin lesions
Bone marrow histology
Bone marrow cytology
Peripheral blood counts
Liver/spleen/lymph nodes
Organ function
Often absent
Multifocal MC infiltrates
MC <20%, low or high grade
Bone marrow may be dys/hyperplastic, but no AHNMD (FAB/WHO)b
Abnormal
Organomegaly
Impaired
Aleukaemic subvariant (in peripheral blood: MC <10%)
Skin lesions
Bone marrow histology
Bone marrow cytology
Peripheral blood counts
Organ function
Absent
‘Positive’ (diffuse and dense)
≥20% MC, often high grade
<10% or ≥10% MC
Impaired (liver, bone marrow, others)
Macroscopic histology
Unifocal, destructive growth
High-grade focal MC tumour
Macroscopic histology
Unifocal, benign tumour
Low-grade focal MC tumour
a Most paediatric cases are diagnosed as CM by skin biopsy and blood examination only; they do not need to have further examinations or tests.
b AHNMD: associated clonal haematological non-mast cell lineage disease; to diagnose an AHNMD, the French-American-British Co-operative Leukaemia Group [85]/World Health Organization [86] (FAB/WHO) criteria have to be fulfilled.
The diagnosis of cutaneous mastocytosis (CM) is based on clinical and histological findings in the skin together with the absence of criteria that would allow the diagnosis of systemic mastocytosis. The criteria for systemic mastocytosis (SM) are divided into major and minor criteria (Box 75.1). Major criteria relate to major histological and immunohistochemical (such as tryptase staining of tissue sections) findings. The most important aspect here is the dense focal infiltrate that consists of a considerable number of MC (>15) and is detectable at more than one site in the tissue(s) (multifocal pattern). Minor criteria relate to typical cytomorphological aspects of MC (in tissue sections and/or bone marrow smears) as well as to novel biochemical markers that show some degree of specificity for SM. It is proposed that if one major and one minor or three minor criteria for SM are fulfilled, the diagnosis is SM. Likewise, if dense multifocal infiltrates in the bone marrow are composed of >15 MC that appear to be spindle-shaped (i.e. where >25% MC are spindle-shaped), the diagnosis of SM can be made on histology without further investigation (and irrespective of the stain applied). If, however, BM infiltrates are subdiagnostic (where there are isolated or small-sized MC infiltrates) or MC are round rather than spindle-shaped, one should seek additional criteria of SM (such as the morphology of MC in bone marrow smears, CD2/CD25 expression on MC, serum tryptase, c-kit mutation) to establish the diagnosis SM.
Box 75.1 Proposed Criteria for Systemic Mastocytosis. Adapted from Valent et al. 2001 [7]
Major
Multifocal dense infiltrates of MC (>15 MC aggregating) detected in sections of bone marrow and/or of other extracutaneous organ(s) by tryptase immunohistochemistry or other stains
Minor
a. >25% of MC in MC infiltrates detected in sections of bone marrow or other extracutaneous organs are spindle-shaped, or presence of >25% atypical MC (type I plus type II)a in bone marrow aspirates
b. Detection of a c-kit point mutation at codon 816 in bone marrow or blood or other extracutaneous organ(s)
c. Kit + mast cells in bone marrow or blood or other extracutaneous organ(s) co-express CD2 and/or CD25
d. Serum total tryptase concentration persistently >20 ng/mL (in case of an associated clonal haematological non-mast cell lineage disease (AHNMO), d. is not valid)b
If one major and one minor, or three minor criteria are fulfilled then the diagnosis is systemic mastocytosis
a See Table 75.4 for morphological criteria and suggested terms.
b In acute myeloid leukaemia, myelodysplastic syndrome or myeloproliferative syndrome, elevated serum tryptase levels have been detected without increase in mast cell numbers or signs of mastocytosis.
Four major variants of SM have been defined by the working group: indolent systemic mastocytosis (ISM), systemic mastocytosis with an associated clonal haematological non-mast cell lineage disease (SM-AHNMD), aggressive systemic mastocytosis (ASM) and mast cell leukaemia (MCL). ISM is characterized by the absence of: (1) signs and symptoms due to impaired organ dysfunction due to infiltration by neoplastic MC; (2) associated clonal haematological non-mast cell lineage disease (AHNMD) (French-American-British (FAB)/WHO criteria), and (3) MCL (no circulating MC, low numbers of immature MC in bone marrow smears). ISM includes isolated bone marrow mastocytosis and smouldering systemic mastocytosis (SSM). In SSM, the clinical course is not aggressive, but clinical and laboratory findings are indicative of slow progression, leading to a huge burden of neoplastic cells (mast cells and/or other myelopoietic cells) over time. These patients present with a hypercellular marrow and organomegaly [10]. AHNMD has to be diagnosed by FAB/WHO criteria.
Aggressive systemic mastocytosis is defined by organ impairment due to MC infiltrates and is characterized by signs and symptoms due to impairment or loss of organ function. In MCL, significant numbers of (immature) MC are detectable in bone marrow (≥20%) and peripheral blood smears. MCL may (first) present as an aleukaemic subvariant (<10% MC in blood smears) or show a leukaemic pattern at diagnosis (MC ≥10% in blood smears).
Mast cell sarcoma (MCS) is characterized by destructive growth of a local tumour consisting of highly atypical (poorly differentiated) MC without systemic involvement (where criteria to diagnose SM are not fulfilled). MCS has to be distinguished from extracutaneous mastocytoma, a localized benign mastocytoma in extracutaneous organs.
Skin disease, with or without systemic involvement, is by far the most common form of childhood mastocytosis. The other variants are rare in children. The authors therefore prefer a more practical classification of childhood mastocytosis for daily use in clinical practice, which divides mastocytosis into cutaneous mastocytosis (with mastocytoma, maculopapular CM and diffuse cutaneous mastocytosis as subclasses), SM and MCL.
The exact prevalence of mastocytosis in the general population is unknown. Children without skin lesions or with indolent cutaneous mastocytosis may remain undiagnosed or unreported. The incidence figures vary widely in various dermatological units from 1 in every 1000 to 1 in 8000 new patients [11,12]. An annual incidence of 5–10 new cases per million population has been estimated [13]. Mastocytosis may occur at any age, but in approximately half of the cases, the onset is between birth and the age of 2 years [13–16]; the disease is congenital in 15%, a further 30% of patients develop mastocytosis before the age of 6 months, another 10% by the age of 2 years, and about 10% between 2 and 15 years of age. There is no clear-cut sex predominance, and children of all races have been affected. Sporadic cases of familial mastocytosis have been reported [16–24].
Aetiology and Pathogenesis
Mast Cells
Mast cells originate from pluripotential haematopoietic bone marrow stem cells that express CD34, CD117 (kit) and CD13 [25]. Final differentiation does not occur in the bone marrow, but morphologically unidentifiable precursor cells leave the bone marrow, migrate into the blood and invade the tissue, where they proliferate and then differentiate into mature mast cells under the control of cytokines [26]. Particularly stem cell factor (SCF, also called mast cell growth factor or kit ligand) appears to play an important role in this proliferation and differentiation process by binding to the human kit protein, the receptor for SCF on the surface of human mast cells [27]. Keratinocytes, endothelial cells, fibroblasts, mast cells and Langerhans cells are the most prominent sources of this cytokine.
Mastocytosis
The remarkable diversity in the clinical presentation of mastocytosis suggests that there are multiple mechanisms contributing to the pathogenesis of disease (Box 75.2) [13]. Discovery of c-kit mutations in patients with SM, however, suggested that the primary pathological process in this disorder is a clonal neoplastic expansion of the cells carrying the mutation [28,29].
Box 75.2 Possible Pathogenetic Mechanisms in Primary Mastocytosis. Modified from Hartmann & Henz 2001 [13]
I. Alterations of the SCF receptor c-kit
A. Activating mutations
B. Inactivating mutations
C. Overexpression (in serum and/or in PBMC of patients with associated haematological disorders)
II. Alterations of mast cell growth factors
A. Increased expression of soluble SCF by keratinocytes (variable)
B. Increased SCF levels in serum (disproven)
C. Increased NGF levels in serum (one patient only)
III. Chromosomal abnormalities
IV. Dysregulation of apoptosis
A. Upregulation of antiapoptotic protein Bcl-2 (in aggressive mastocytosis)
B. Upregulation of antiapoptotic protein Bcl-X (in bone marrow of indolent mastocytosis)
SCF, stem cell factor; PBMC, peripheral blood mononuclear cells; NGF, nerve growth factor.
c-kit Mutations.
The SCF receptor (kit) is encoded by the proto-oncogene c-kit and belongs to the type III transmembrane receptor tyrosine kinase subfamily [26] (Fig. 75.1). Apart from MC and their progenitors, kit is also expressed on other haematopoietic and non-haematopoietic (progenitor) cells [30]. Ligation of kit by SCF induces receptor dimerization followed by transphosphorylation of tyrosine residues becoming docking sites for the recruitment and activation of various cellular substrates. The activated substrates then induce multiple intracellular signalling pathways responsible for MC differentiation, proliferation, survival and activation [31,32].
Fig. 75.1 Schematic representation of the structure of the human c-kit receptor and location of the main point mutations found in patients with childhood-onset mastocytosis
Adapted from Yanagihori et al. 2005 [40].
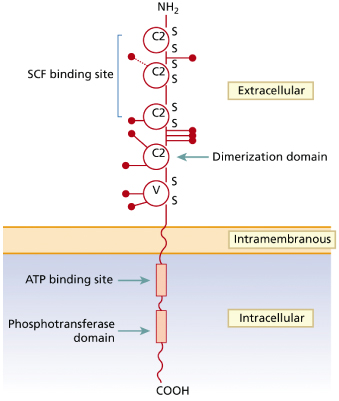
With regard to non-MC diseases, c-kit mutations have been described in a subset of acute myeloid leukaemias (AML), myeloproliferative syndromes, sinonasal lymphomas and gastrointestinal stromal tumours [28,33].
A number of studies have investigated the role of SCF and kit in the pathogenesis of mastocytosis.
Earlier observations raised the possibility of an altered metabolism of SCF in some patients with CM [34]. However, such a mechanism may not apply to the majority of patients. Screening for abnormalities in the sequence of c-kit revealed somatic mutations in the c-kit gene in rodent and human MC lines. Many of these mutations are associated with kit phosphorylation and downstream activation, independent of SCF binding [35].
These activating c-kit mutations can be classified into two major groups based on their topological localization [33]. The ‘regulatory type’ mutations typically affect regulation of the kinase activity of the kit molecule by disrupting the auto-inhibitory α-helix at the juxtamembrane domain of kit. These mutations affect the binding of signal transducing or regulatory molecules to kit inducing ligand-independent dimerization and activation; most frequently, these ‘regulatory type’ mutations occur at the juxtamembrane domain of kit [36]. The second type are the ‘enzymatic pocket’ mutations which directly affect the enzymatic site at the TK2 activation loop and induce activation of kit in the absence of dimerization of the receptor [33].
Based on cell line models, mutations of c-kit have been investigated and described in various forms of human mastocytosis (Table 75.2) [28]. Among the mutations found, the most common is the mutation Asp to Val at codon 816 (c-kitD816V) [33]. Typical paediatric mastocytosis patients with indolent disease lack c-kitD816V mutations [37–39], although a recent study of Japanese patients found that 12 of 14 patients with childhood-onset cutaneous mastocytosis had D816 substitutions [40]. Surprisingly, a point mutation of codon 839 with substitution of lysine for glutamic acid (c-kitE839K) was found by Longley et al. [38] in three of six paediatric cases of CM. Interestingly, c-kitE839K was neither autophosphorylated nor phosphorylated after exposure to SCF. Thus, this mutation appeared to be an ‘inactivating’ mutation.
Table 75.2 Known c-kit mutations in mastocytosis.
Adapted from Orfao et al. 2007 [28]
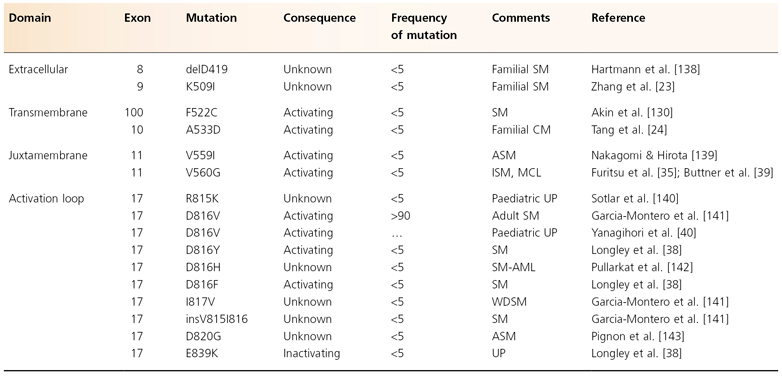
CM, cutaneous mastocytosis; SM, systemic mastocytosis; AML, acute myeloblastic leukaemia; ISM, indolent systemic mastocytosis; UP, urticaria pigmentosa (= maculopapular mastocytosis); CML, chronic myeloid leukaemia; MCL, mast cell leukaemia; MF, myelofibrosis; MPD, myeloproliferative disorder; ASM, aggressive systemic mastocytosis; WDSM, well-differentiated systemic mastocytosis.
Based on c-kit mutations, mastocytosis can be divided into three major groups [26].
The first presents with ‘activating’ c-kit mutations, mainly at codon 816. This group includes most cases of ISM, but also cases of SM-AHNMD, ASM and MCL, as well as some (atypical possibly persisting and later systemic) paediatric cases [37]. It remains unknown, however, why patients bearing the same activating mutation exhibit variable clinical courses ranging from indolent to highly aggressive. c-kit mutation may involve the mast cell progenitors at different levels of commitment with varying potentials for expansion. The c-kitD816V mutation may affect a committed MC progenitor in patients with limited indolent disease. In contrast, patients with more extensive disease variants, such as smouldering SM or aggressive mastocytosis, may have an earlier pluripotential progenitor cell affected, resulting in multilineage haematopoietic involvement similar to other myeloproliferative disorders [28,41–43]. The relatively stable course of the disease in most SM patients and the observation that the same c-kit mutation (e.g. c-kitD816V) may be associated with indolent (good prognosis) and malignant tumours may underline the potential role of other genetic and/or epigenetic factors in determining the progression/outcome of the disease [28]. It has been suggested [44] that c-kitD816V alone might affect the differentiation and apoptosis potential of human MC and is sufficient to cause indolent SM with a favourable course. Additional defects may be required to cause severe types of SM by causing proliferation [45].
In very few patients (with CM) ‘inactivating’ mutations may be found, but the significance of this finding is unknown.
In a third group of patients no c-kit mutations can be found. These cases include the majority of typical paediatric CM, a subset of ASM and MCL, and the majority of the rare cases of familial mastocytosis [38]. It is not clear whether in these patients no mutation or a so far unknown mutation has happened [22–24,46].
Alterations of Mast Cell Growth Factors.
One study identified an increased local concentration of soluble SCF in lesions of human cutaneous mastocytosis [34]. More extended studies have, however, been unable to confirm these results [47,48]. Furthermore, soluble c-kit protein is elevated in the serum of indolent SM, mastocytosis with an associated haematological disorder and aggressive mastocytosis, compared with maculopapular CM without systemic involvement and healthy controls [49]. This observation is most likely to be due to an increase of c-kit-bearing MC precursors in the blood of these patients.
Other factors, such as nerve growth factor, also may be involved in the pathogenesis of mastocytosis [27,50–53]. Platelet-derived growth factor (PDGF) is another important cytokine for growth and survival of human mast cells although its effects are not restricted to MC lineage [54].
Apoptosis of Mast Cells.
Cervero et al. [55] observed a strongly enhanced expression of the antiapoptotic protein Bcl-2 in mast cells from the bone marrow of one patient with MCL. In comparison, there was no increased expression of Bcl-2 in patients with indolent mastocytosis, paediatric mastocytosis or reactive mast cell hyperplasia due to other diseases. Likewise, Baldus et al. [44] found altered apoptosis and cell cycling of mast cells in bone marrow lesions of patients with SM. This suggests that some forms of mastocytosis may also be associated with the inhibition of apoptosis, the physiological form of cell death. Using immunohistology, bone marrow mast cells of mastocytosis patients have also been found to express the antiapoptotic protein Bcl-X [56].
Chromosomal Abnormalities.
The rate of chromosomal abnormalities appears to be increased in patients with mastocytosis, especially in those with an associated haematological disorder [57–60]. The affected chromosomal regions, however, fail to correspond to genes that may be related to the pathogenesis of mastocytosis, such as c-kit, interleukin (IL)-4, IL-6 or IL-9. Worobec et al. therefore suggested that chromosomal abnormalities in mastocytosis patients may not directly be associated with c-kit mutations, but both defects could result from a common altered repair mechanism [57].
Phenotype of Mastocytosis Mast Cells.
Detailed studies that clearly demonstrate an increased proliferation of mast cells in patients with mastocytosis are still lacking. Immunohistochemical analyses of mast cells in maculopapular CM and mastocytoma skin lesions for mast cell and monocytic markers have so far also not yielded any unusual features, except for a somewhat immature phenotype in mastocytomas [61,62]. Recent analyses of bone marrow mast cells from mastocytosis patients by immunohistochemistry and multiparameter flow cytometry showed, however, that mast cells in mastocytosis express more CD2 than controls, a receptor normally restricted to T and natural killer cells [63,64]. In addition, the cells express CD25, which is usually expressed on activated T-cells [65,66] Further studies have now proved that CD25 is a reliable immunohistochemical marker for the discrimination of neoplastic from normal/reactive MC in SM, irrespective of the SM subtype [67], while screening for CD2 has been shown to have lower diagnostic value because a significant proportion of cases stain negative. Besides, CD2 expression on bone marrow MC is generally weak in the cases that are positive [67–69].
Mast Cell Mediators
The mast cell has the capacity to generate a wide variety of mediators that are responsible for both immediate and long-term effects on their target organ. The major mast cell secretory products are shown in Box 75.3. The mast cell secretes its pharmacological agents in response to an appropriate pathophysiological stimulus by a process resembling exocytosis [70]. There are a number of well-recognized activators of mast cell secretion, both immunological and non-immunological [71, 72] (Box 75.4).
Box 75.3 Human Mast Cell Products. Adapted from Longley et al. 1995 [137]
Secretory Granule Preformed Mediators
• Histamine
• Proteoglycans (heparin, chondroitin sulphate)
• Neutral proteases (tryptase, chymase, cathepsin G, carboxypeptidase)
• Acid hydrolases (β-hexosaminidase, β-glucuronidase, arylsulphatase, N-acetyl-β-glucosaminidase)
Lipid Mediators
• Leukotriene C4
• Platelet-activating factor
• Prostaglandin D2
Cytokines
• Interleukin 4, 5, 6 and 8
• Tumour necrosis factor-α
Box 75.4 Clinically Relevant Mast Cell Degranulators. Modified from DiBacco & DeLeo 1982 [72] and Stein 1986 [71]
- Immunological stimuli (IgE)
- Complement-derived anaphylotoxins (C3A and C5A)
- Physical stimuli (cold, heat, sunlight, friction)
- Polymers (compound 48/80; dextran)
- Bacterial toxins
- Snake venoms
- Hymenoptera venoms
- Biological polypeptides (released by Ascaris, jellyfish, crayfish and lobster)
- Drugs
- acetylsalicylic acid
- alcohol
- narcotics (e.g. codeine, morphine)
- procaine
- polymyxine B
- amphotericin B
- atropine
- thiamine
- D-tubocurarine
- quinine
- radiographic contrast media containing iodine
- scopolamine
- gallamine
- decamethonium
- reserpine
- acetylsalicylic acid
Pathology.
Depending on the organ system analyzed, the shape of the mast cells, the maturation stage of the mast cells, the type of mast cell disease and presence of other (concomitant) disorders, it may be difficult to diagnose mastocytosis. Routine formalin fixation and toluidine blue stains are often sufficient for diagnostic purposes but in doubtful cases, immunohistochemical techniques using antibodies against mast cell-associated antigens such as tryptase, CD2, CD25 and CD117 (kit) are helpful.
Irrespective of the clinical type of mastocytosis, biopsy of lesional skin shows significantly increased numbers of spindle-shaped mast cells in the dermis [73–76] (Fig. 75.2
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


