7 Blepharoptosis
Abstract
“Blepharoptosis” discusses drooping of the upper eyelid (the term ptosis refers to the drooping of any body part). A ptosis can affect all age groups and be congenital or acquired. The causes of ptosis are numerous. It is important to appreciate that ptosis itself is merely a physical sign, and not a diagnosis. It is therefore essential to determine the underlying cause before any decision is made about the surgical management of a patient with a ptosis. A ptosis can be the presenting sign of a serious or life-threatening systemic disorder. Additionally, pseudoptosis is a condition that mimics a true ptosis. It is particularly important to differentiate a pseudoptosis clearly from a true ptosis and to exclude a neurological or myogenic cause of ptosis, requiring further evaluation or an alternative therapeutic approach, before undertaking ptosis surgery.
7.1 Introduction
The term ptosis refers to the drooping of any body part (e.g., eyebrow ptosis, midface ptosis, lash ptosis). In this chapter, the term will be used to refer to a drooping of the upper eyelid (a blepharoptosis). A ptosis is commonly encountered by all ophthalmic surgeons, irrespective of their area of interest. It can affect all age groups and may be one of the following:
Congenital.
Acquired.
The causes of ptosis are numerous. It is important to appreciate that ptosis itself is merely a physical sign, and not a diagnosis. It is therefore essential to determine the underlying cause before any decision is made about the surgical management of a patient with a ptosis. It is particularly important to bear in mind that a ptosis can be the presenting sign of a serious life-threatening systemic disorder (Fig. 7‑1).
In considering the causes, it is useful to use a classification of ptosis which is based upon etiological factors (Box 7.1). This should be borne in mind when examining a patient who presents with a ptosis.
Box 7.1 Classification of Ptosis
Pseudoptosis
True ptosis
Neurogenic
Myogenic
Aponeurotic
Mechanical
Key Point
A ptosis can be the presenting sign of a serious life-threatening systemic disorder.
7.2 Classification
The classification of ptosis aims to provide some insight into the pathological processes involved. A ptosis can also be classified as one of the following:
Pseudoptosis.
True ptosis.
A pseudoptosis should be differentiated from a true ptosis. A true ptosis, in turn, can be categorized as neurogenic (caused by an abnormality of the innervation of the levator palpebrae superioris or of Müller’s muscle), myogenic (caused by an abnormality of the levator muscle itself), aponeurotic (caused by a defect within the levator aponeurosis or in the attachment of the aponeurosis to the tarsus), or mechanical (caused by the mechanical effects of adhesions in the superior conjunctival fornix or of a mass in the upper eyelid and/or anterior superior orbit). These etiological mechanisms occur in all age groups but with varying frequency. Congenital “dystrophic” ptosis and involutional aponeurotic ptosis are by far the most common types of ptosis encountered in clinical practice.
7.2.1 Pseudoptosis
Pseudoptosis (Box 7.2) refers to a condition that mimics a true ptosis.
Box 7.2 Pseudoptosis
Contralateral eyelid retraction
Hemifacial spasm
Aberrant reinnervation of the facial nerve
Postenucleation socket syndrome
Double elevator palsy
Dermatochalasis/brow ptosis
Duane’s retraction syndrome
Key Point
It is particularly important that a pseudoptosis is clearly differentiated from a true ptosis and that a neurological, myogenic, or mechanical cause of ptosis requiring further evaluation or an alternative therapeutic approach is excluded before ptosis surgery is embarked upon.
Contralateral Eyelid Retraction
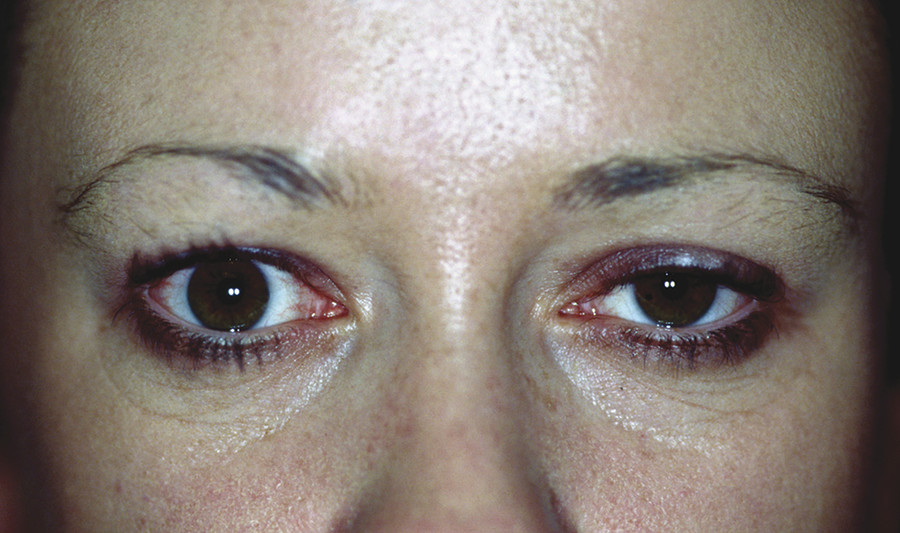
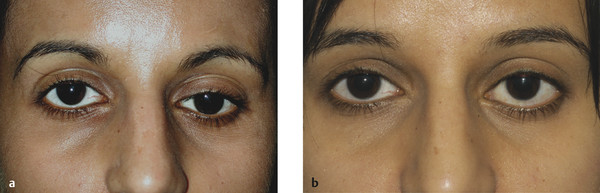
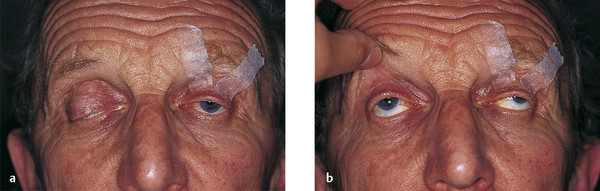
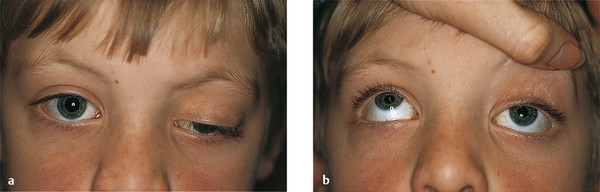
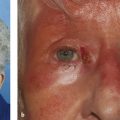
Contralateral upper eyelid retraction may lead to diagnostic confusion, with the normal eyelid appearing to be ptotic. A patient presenting with unilateral eyelid retraction (e.g., from undiagnosed thyroid eye disease) may complain of a ptosis affecting the fellow eyelid (Fig. 7‑2).
It is also important to differentiate contralateral upper eyelid retraction resulting from the patient exerting maximum levator effort on the side of a true ptosis. This is particularly prevalent when the ptosis affects the dominant eye.
Hemifacial Spasm
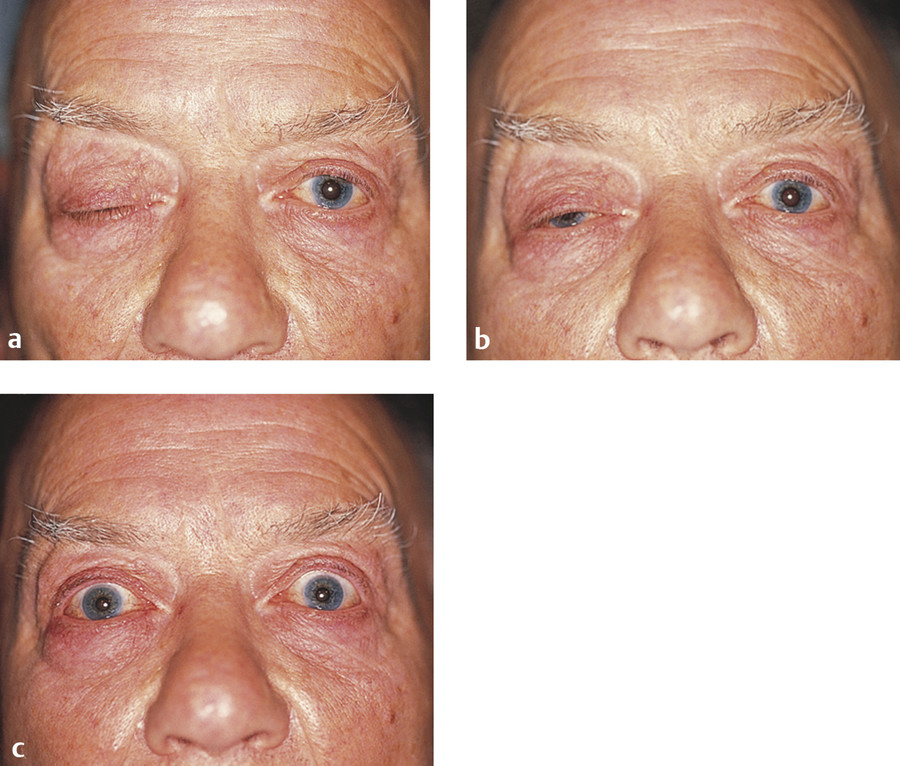
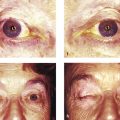
Hemifacial spasm is characterized by unilateral, involuntary, intermittent, and irregular contractions of the muscles of facial expression (Fig. 7‑3). The orbicularis oculi muscle is usually the first facial muscle to be involved. In some patients the early signs may be quite subtle. The patient will typically complain of a droopy eyelid, but, in addition to an apparent blepharoptosis, the position of the lower eyelid will be seen to be higher than the fellow eyelid, causing an even greater vertical narrowing of the palpebral aperture. In some cases, the cause is compression of the facial nerve in the posterior cranial fossa by an aberrant artery. In these cases, neurosurgical decompression of the nerve may be successful in controlling the spasms when they are severe, but the risks of such invasive surgical intervention should be considered very carefully. Injections of botulinum toxin (e.g., 2–3 units of Azzalure per injection) into the medial and lateral aspects of the upper and lower eyelids can be very successful in controlling the associated blepharospasm but may lead to significant orbicularis oculi muscle weakness with the need to use very frequent topical lubricants to prevent exposure keratopathy. In addition, such injections are associated with the risk of diplopia from extension of the toxin to the inferior oblique muscle, and with the risk of a true blepharoptosis from extension of the toxin to the levator palpebrae superioris muscle.

Aberrant Reinnervation of the Facial Nerve
Aberrant reinnervation of the facial nerve may occur after a peripheral lower motor neuron facial nerve palsy. This condition is characterized by involuntary eyelid closure induced by the use of other facial muscles, such as on smiling or whistling (Fig. 7‑4). The involuntary eyelid closure may be controlled by local injections of botulinum toxin as described previously. Ptosis surgery is usually contraindicated in this condition.
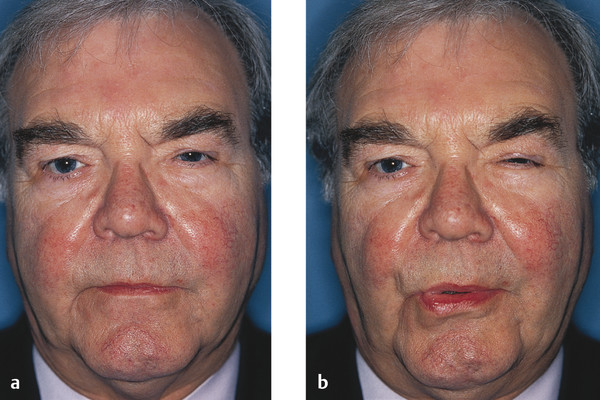
Key Point
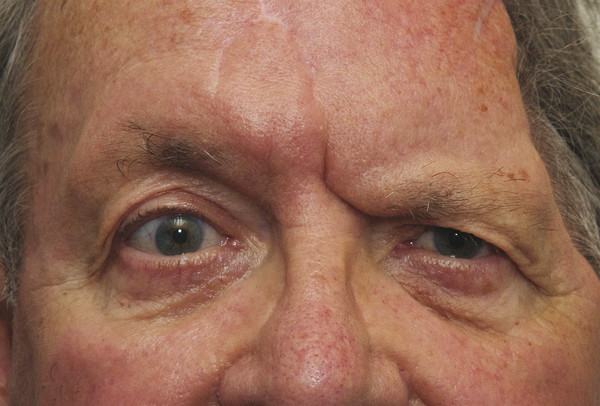
It is important to exclude the possibility of aberrant regeneration in any patient referred with an apparent ptosis who gives a prior history of a facial palsy. Patients with a prior history of a facial palsy are also likely to have a brow ptosis, which can also simulate a ptosis (Fig. 7‑5).
Postenucleation Socket Syndrome
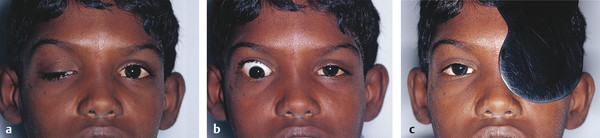
Ptosis (or indeed eyelid retraction in some patients) may be seen as one of the features that typify a postenucleation socket syndrome (Fig. 7‑6). It is believed to be related to the loss of the fulcrum for the action of the levator palpebrae superioris. This is corrected by addressing a socket volume deficit with the insertion of an orbital implant and by the provision of a correctly fitting ocular prosthesis. (A true ptosis may also exist in such patients). A similar situation is seen in a patient with an orbital volume expansion after an untreated orbital floor blowout fracture.
Hypotropia
A ptosis that occurs in conjunction with a hypotropic eye (e.g., a double elevator palsy) may resolve completely once the eye has been repositioned surgically, usually using a Knapp procedure (Fig. 7‑7).
The Knapp procedure is useful for treating a double elevator palsy, especially when there is no mechanical restriction to elevation. This surgical technique uses an upward shift of the medial and lateral rectus muscles to a point adjacent to the borders of the insertion of the superior rectus muscle.
A cover test should be performed on all patients presenting with a history of a congenital ptosis to exclude the possibility of such a pseudoptosis (Fig. 7‑8).
Dermatochalasis and Brow Ptosis
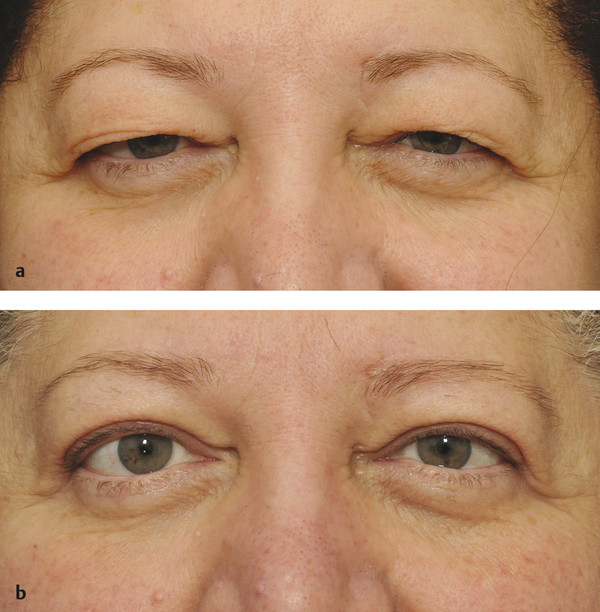
Dermatochalasis (an age-related laxity and redundancy of upper eyelid skin) and/or a brow ptosis may mimic an upper lid ptosis that resolves once a blepharoplasty or a browlift has been performed (Fig. 7‑9).
Duane’s Retraction Syndrome
Narrowing of the palpebral fissure may be associated with horizontal ocular movements. Patients may present with a complaint of ptosis, but a careful examination of the patient’s ocular motility reveals the true diagnosis (Fig. 7‑10).
7.2.2 True Ptosis
Neurogenic Ptosis
For a classification of neurogenic ptosis, see Box 7.3.
Box 7.3 Neurogenic Ptosis
Oculomotor nerve palsy
Horner’s syndrome
Myasthenia gravis
Synkinetic ptosis
Marcus-Gunn jaw-winking phenomenon
Aberrant reinnervation of the oculomotor nerve
Guillain–Barré syndrome
Cerebral ptosis
Spread of botulinum toxin to the levator muscle after periocular injection
Botulism
Oculomotor Nerve Palsy (Third Cranial Nerve Palsy)
Clinical Features
A third nerve palsy is characterized by a variable degree of ptosis associated with deficits of adduction, elevation, and depression of the eye related to weakness of the levator palpebrae superioris muscle; the superior, inferior, and medial rectus muscles; and the inferior oblique muscle (Fig. 7‑11). The pupillary fibers of the third cranial nerve may be affected or spared depending on the underlying cause. Lesions confined to the superior division of the third nerve result in a ptosis and weakness of the superior rectus muscle only. The patient’s Bell’s phenomenon is typically absent or poor. Myasthenia may mimic a third nerve palsy when the pupil is spared, and this should always be considered.
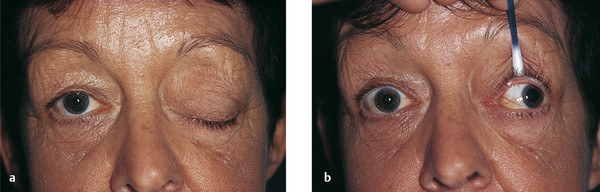
Cyclic oculomotor nerve palsy is a rare phenomenon characterized by alternating paresis and spasm of the extraocular and intraocular muscles. These cyclic phenomena are usually noted in early childhood and may be evident at birth.
Etiology
A third nerve palsy may be caused by the following:
Neoplastic lesions.
Inflammatory lesions.
Vascular lesions.
Traumatic lesions.
Any of these lesions may affect the nerve in its course from the midbrain to the orbit. Associated symptoms and signs help to localize the underlying lesion. Damage to the third nerve within the subarachnoid space produces an isolated third nerve palsy.
The main vascular cause of a third nerve palsy is compression of the nerve by an expanding aneurysm of the posterior communicating artery or the basilar artery.
There is commonly pain with aneurysmal compression, and pupillary involvement is typical, although there have been occasional cases of aneurysmal compression that did not initially affect pupillary function. In ischemic vascular third nerve palsies, pain is uncommon and the pupil is typically normal and reactive. The majority of ischemic vascular third nerve palsies resolve spontaneously and show a gradual recovery over a period of 3 to 6 months.
Damage to the third nerve in the cavernous sinus, superior orbital fissure, or posterior orbit is unlikely to present as an isolated third nerve palsy because of the confluence of other structures in these areas.
Cavernous sinus involvement may also include pareses of the fourth and sixth cranial nerves, the ophthalmic division of the fifth nerve, and an ipsilateral Horner’s syndrome. The most common causes of damage to the third nerve in these areas include the following:
Metastatic disease.
Orbital inflammatory disease.
Herpes zoster.
Carotid artery aneurysm.
Pituitary adenoma.
Pituitary apoplexy.
Sphenoid wing meningioma.
In complicated third nerve palsies where other neural structures are involved, the patient should undergo magnetic resonance imaging (MRI). In isolated third nerve palsies with no pupillary involvement, when the patient is older than 50 years, MRI scanning, investigations for the cause of a vasculopathy, and a daily pupillary evaluation are indicated.
Investigation
For patients younger than 50 years presenting with an isolated pupil-sparing third nerve palsy, intracranial angiography is indicated, because an ischemic vasculopathy is far less likely to occur in this age group than is an aneurysm. If an adult patient of any age presents with a complete or incomplete isolated third nerve palsy with pupillary involvement, this should be considered to be a medical emergency, and the patient should undergo urgent intracranial angiography. In these cases, the cause is likely to be a life-threatening subarachnoid aneurysm. Most third nerve palsies presenting in children are traumatic or congenital.
Key Point
If an adult patient of any age presents with a complete or incomplete isolated third nerve palsy with pupillary involvement, this should be considered to be a medical emergency, and the patient should undergo urgent intracranial angiography.
Management
Treatment of the ptosis associated with a third nerve palsy is problematic because of the impaired Bell’s phenomenon with a risk of exposure keratopathy. A frontalis suspension procedure may be undertaken after strabismus surgery has been performed to realign the globe in the primary position. A frontalis suspension procedure may be undertaken before strabismus surgery in infants to treat amblyopia. It is wise to use a 4–0 polypropylene suture for the frontalis suspension, because it can easily be removed and the procedure reversed if the patient develops exposure keratopathy.
Key Point
Myasthenia gravis has the ability to mimic virtually any cranial neuropathy, including isolated pupil-sparing third nerve palsies. Myasthenia gravis must remain a possible diagnosis when encountering a third nerve palsy if the clinical course is variable or atypical.
Horner’s Syndrome (Oculosympathetic Paresis)
Horner’s syndrome is a rare condition that results from a disruption of the sympathetic nerve supply to the head and neck. In France this is commonly referred to as Bernard–Horner’s syndrome.
Clinical Features
Horner’s syndrome is characterized by the following:
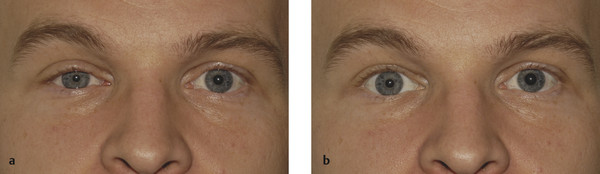
A ptosis of 1 to 2 mm with good levator function and a raised skin crease.
A pupillary miosis.
An apparent enophthalmos.
It is occasionally associated with facial anhidrosis and facial flushing depending on the site of the lesion. The innervation of sweat glands in the medial and lateral parts of the forehead is also different, the medial part being supplied by nerve fibers from the sympathetic plexus of the internal carotid artery, while the sweat glands in the lateral part are furnished from the plexus surrounding the external carotid artery.
The eyelid features are related to interference with the sympathetic nerve supply to Müller’s muscle in the upper eyelid and to its smooth muscle counterpart in the lower eyelid, and to the dilator pupillae muscle. The resultant anisocoria is accentuated in dim illumination. The apparent enophthalmos is caused by the decrease in the vertical height of the palpebral aperture (Fig. 7‑12). Iris heterochromia may also be seen if the Horner’s syndrome occurs before 2 years of age. Iris pigmentation, which is under sympathetic control during early development, is completed by the age of 2 years, making heterochromia an uncommon finding in Horner’s syndrome acquired later in life. Old photographs can aid the clinician in distinguishing congenital Horner’s syndrome from acquired Horner’s syndrome by documenting heterochromia present at, or close to, birth.
Neuroanatomy
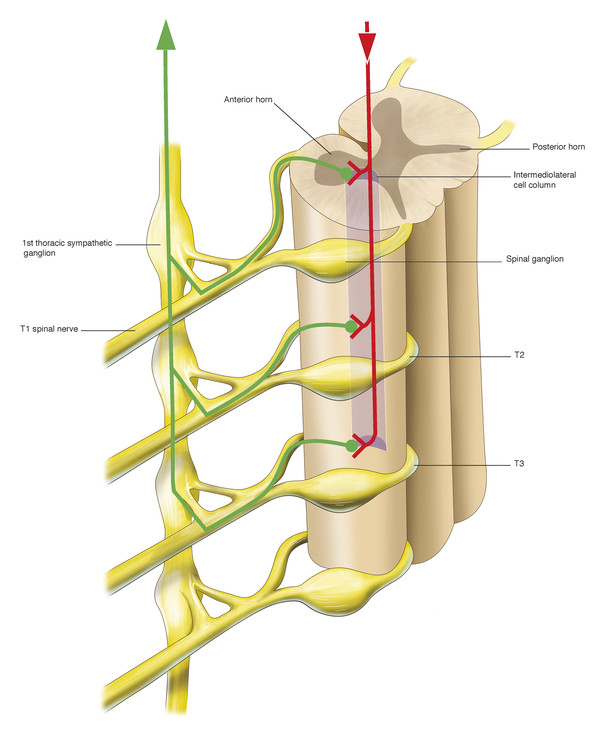
Horner’s syndrome may be caused by a lesion that interrupts the course of the sympathetic neurons anywhere from their origin in the hypothalamus to the orbit (Fig. 7‑13).
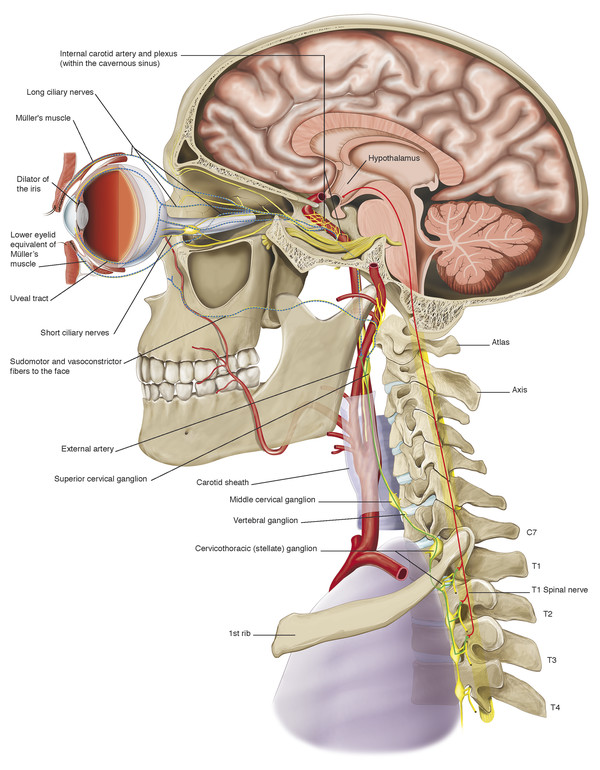
There are three orders of neuron carrying the sympathetic innervation to the orbit. The first neuron commences in the hypothalamus and descends and travels between the levels of the eighth cervical and fourth thoracic vertebrae (C8–T4) of the spinal cord, synapsing with the second neuron in the intermediolateral cell column (Fig. 7‑14).
There, the preganglionic cell bodies of the second order neurons give rise to axons. These axons pass over the apex of the lung and across the neck of the first rib and then ascend behind the carotid sheath, entering the sympathetic chain in the neck (Fig. 7‑14). They synapse with the third neuron in the superior cervical ganglion, which lies in front of the lateral mass of the atlas and axis. There, cell bodies of third order neurons give rise to postganglionic axons that travel to the eye via the cavernous sinus. Sympathetic nerves enter the orbit through the superior orbital fissure via the first and second divisions of the trigeminal nerve and a plexus of nerves surrounding the ophthalmic artery. These sympathetic nerve fibers course anteriorly through the uveal tract and join the fibers of long posterior ciliary nerves to innervate the dilator of the iris, the dilator pupillae muscle. Postganglionic sympathetic fibers also innervate Müller’s muscle in the upper eyelid and its smooth muscle equivalent in the lower eyelid after travelling anteriorly in the orbit via the nasociliary and lacrimal nerves. Postganglionic sympathetic fibers, responsible for facial sweating, follow the external carotid artery and its branches to the sweat glands and blood vessels of the neck, face, and head. Interruption at any location along this pathway (preganglionic or postganglionic) will induce an ipsilateral Horner’s syndrome.
Etiology
The causes of an acquired central (first order neurone) Horner’s syndrome include the following:
Cerebrovascular accidents.
Multiple sclerosis.
Basal skull tumors.
Basal meningitis.
Neck trauma (e.g., vertebral artery dissection).
Syringomyelia.
Arnold–Chiari malformation.
Spinal cord tumors.
The causes of acquired preganglionic (second order neuron) Horner’s syndrome include the following:
Trauma.
Aortic dissection.
Carotid artery dissection.
Tuberculosis.
Apical lung tumors (e.g., Pancoast tumor).
Lymphoma.
Mediastinal tumors.
Tuberculosis.
Lower brachial plexus trauma.
Cervical rib.
Aneurysms of the aorta or subclavian or common carotid arteries.
Neck or chest surgery.
Neuroblastoma.
The causes of a postganglionic (third order neurone) Horner’s syndrome include the following:
Cluster migraine headache.
Herpes zoster infection.
Internal carotid artery dissection.
Raeder’s syndrome (paratrigeminal syndrome).
Caroticocavernous fistula.
Important Points
Isolated Horner’s syndrome is typically vascular in nature.
Horner’s syndrome associated with pain needs to be investigated. If there is arm, shoulder, or hand pain, a Pancoast’s tumor should be considered. If a patient experiences pain in the face or neck, a carotid artery dissection should be considered. A combination of pain and amaurosis fugax may be related to carotid dissection.
Unless there is a known etiology such as birth trauma from a forceps delivery, all children with acquired Horner’s syndrome should be investigated, because there is commonly a serious underlying disorder such as a neuroblastoma.
Preganglionic lesions are less common but more likely to be malignant.
Investigations
Investigations should be undertaken depending of the suspected underlying cause:
Chest radiography may show an apical carcinoma of the lung.
Computed tomography (CT) or MRI may be necessary to identify a cerebrovascular accident.
Angiography or ultrasonography may be necessary to demonstrate a dissection of the carotid artery.
Diagnostic Tests
The diagnosis of Horner’s syndrome is made clinically but may be confirmed with pharmacological testing by the instillation of 5% cocaine solution into both eyes. Cocaine acts as an indirectly acting sympathomimetic agent by inhibiting the reuptake of noradrenaline at the nerve ending. A Horner’s pupil, in contrast to a normal pupil, will fail to dilate, or will dilate poorly, because of the absence of endogenous noradrenaline at the nerve ending. The pupils should be evaluated 30 minutes after the instillation of the drops to ensure accuracy. The cocaine test is used to confirm or rule out Horner’s syndrome. A positive cocaine test does not, however, localize the lesion. If cocaine is not available or if there may be a parental objection to its use in a child, an alternative option is the use of 1% apraclonidine (Lopidine, Novartis Pharmaceuticals UK Ltd.), an α-adrenergic receptor agonist.
To localize the lesion as either preganglionic or postganglionic, 1% hydroxyamphetamine solution can be instilled into the eye after waiting for a further 48 hours. (It is important to delay this test, because cocaine can inhibit the uptake of hydroxyamphetamine into the presynaptic vesicle). Hydroxyamphetamine displaces noradrenaline from the presynaptic vesicles. If the third neuron is damaged, there will be no endogenous noradrenaline and the pupil will not dilate, thus indicating a postganglionic lesion. Dilation indicates a first-order or a second-order neuron lesion. There is currently no method of pharmacological testing that can differentiate a first-order preganglionic lesion from a second-order preganglionic lesion. The differentiation between a first-order and a second-order neural lesion is based on the neurological signs associated with a first-order neural lesion.
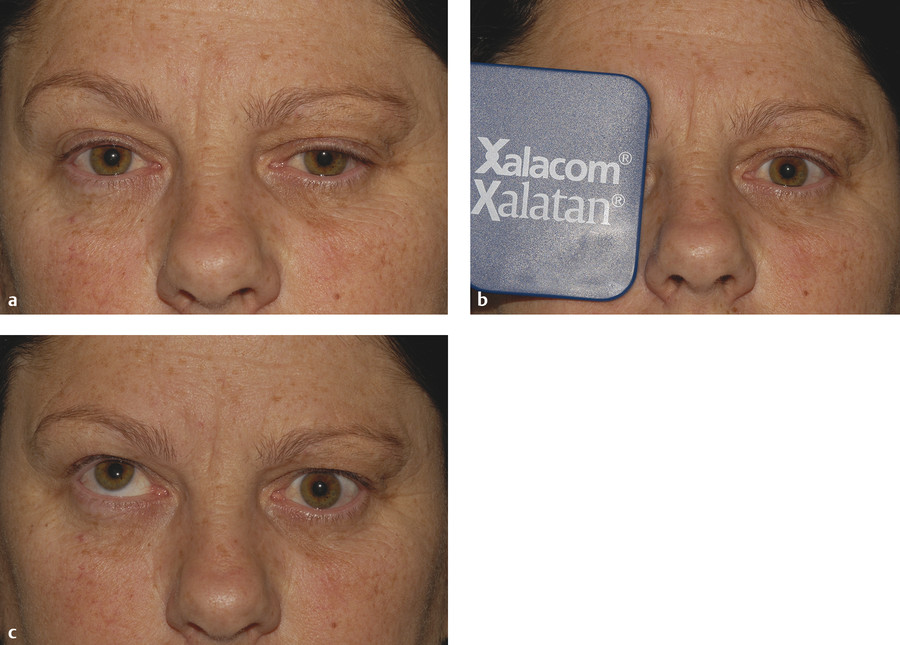
The instillation of a weak solution of phenylephrine (1%) may demonstrate denervation hypersensitivity, resulting in a temporary resolution of the ptosis and restoration of a normal skin crease (Fig. 7‑12). It may also restore the lower lid to its normal position.
Key Point
It is important to differentiate preganglionic from postganglionic Horner’s syndrome, because lesions that result in postganglionic Horner’s syndrome are usually benign (usually vascular in nature), in contrast to those resulting in preganglionic Horner’s syndrome.
In general, the treatment for Horner’s syndrome depends upon the underlying cause. In many cases there is no treatment that improves or reverses the condition. Treatment in acquired cases is directed toward eradicating the lesion that is responsible for the syndrome.
The ptosis may be treated surgically either by means of a posterior-approach Müller’s muscle resection or by means of a levator aponeurosis advancement procedure. It is very difficult to improve the slightly elevated position of the lower eyelid surgically. I believe a posterior-approach Müller’s muscle resection is the surgical procedure of choice for the management of ptosis in a patient with Horner’s syndrome.
Key Point
A posterior-approach Müller’s muscle resection is the surgical procedure of choice for the management of ptosis in a patient with Horner’s syndrome.
Myasthenia Gravis
Myasthenia gravis is an autoimmune disorder caused by antibodies to the acetylcholine receptors of the motor endplate of voluntary muscles. The antibodies block access of the neurotransmitter acetylcholine to the receptors. The hallmarks of the disorder are variable muscular weakness and fatigue on exercise. Myasthenia may be generalized and may threaten the muscles of respiration (myasthenia gravis), or it may be localized to the eyes (ocular myasthenia).
Clinical Features
Approximately 30% of patients present with ocular signs and symptoms (ptosis and diplopia), whereas 80% to 90% of patients have ocular signs at the time of diagnosis. Thyroid disorders may be seen in as many as 10% of patients with myasthenia gravis, and symptoms of hyperthyroidism or hypothyroidism may also be present.
If the symptoms and signs remain confined to the eyes for 3 years, progress to generalized myasthenia gravis is unlikely. Ptosis is the most common clinical manifestation of myasthenia. It may be unilateral or bilateral. Exercise of the levator palpebrae superioris muscle or sustained upgaze may provoke or worsen a ptosis. Attempted rapid saccades from downgaze to the primary position may provoke an overshoot of the upper eyelid above the superior limbus with a gradual fall of the lid to its original position (Cogan’s twitch sign). This is not, however, pathognomonic. There may be an associated weakness of the orbicularis oculi muscle, and the Bell’s phenomenon may be poor.
Diagnostic Tests
The possible diagnosis of myasthenia should be contemplated in any patient with an acquired ptosis and normal pupils. The diagnosis may be confirmed by means of a Tensilon test (edrophonium chloride) (Fig. 7‑15). Tensilon is a short-acting anticholinesterase agent that increases the amount of acetylcholine available at the motor endplate when administered intravenously. In most cases it will temporarily and dramatically overcome the muscle weakness of myasthenia. Failure to do so, however, does not exclude the diagnosis. Precautions should be taken before performing the Tensilon test. Resuscitation equipment should be available and an intravenous cannula placed for venous access, because, very occasionally, edrophonium can cause a significant bradycardia, heart block, and asystole. Vital signs should be monitored before and during the test. Atropine should be drawn up and ready to administer in the event of the development of any of these adverse systemic events. A small intravenous test dose (2 mg) of Tensilon should be given and the response observed before injection of a further 8 mg to ensure there are no adverse side effects.
Other confirmatory tests can be performed, such as an acetylcholine receptor antibody assay, standard electromyography, single-fiber electromyography, and repetitive nerve stimulation electromyography to evaluate decremental responses.
A very simple clinical test is the ice pack test. Cooling may improve neuromuscular transmission. In a patient with myasthenia gravis who has ptosis, placing crushed ice in a surgical glove over the eyelid for 2 minutes can lead to improvement of the ptosis.
Management
Treatment of a patient with myasthenia should be undertaken by a neurologist. The treatment may involve the use of anticholinesterase agents, systemic steroids, immunosuppressants, or plasmapheresis. CT or MRI of the chest is highly accurate in detecting a thymoma. Every patient with myasthenia gravis should be screened for a thymoma. Thymectomy may be beneficial in some cases.
The ophthalmologist plays a role in the management of ptosis or diplopia unresponsive to medical therapy. The ptosis may be treated by the use of ptosis props if the Bell’s phenomenon is absent and if the orbicularis oculi muscle function is poor. Patients with normal orbicularis oculi muscle function rarely tolerate ptosis props.
The surgical management depends on the levator function. If this is better than 4 to 5 mm, an anterior or posterior levator aponeurosis advancement procedure may be used. If the levator function is less than 4 mm, a frontalis suspension procedure will be necessary. The risk of inducing exposure keratopathy must be considered carefully before embarking on such surgery.
Key Point
The possible diagnosis of myasthenia should be contemplated in any patient with an acquired ptosis and normal pupils.
Synkinetic Ptosis
A synkinesis is simultaneous movement of muscles supplied by different nerves or by separate branches of the same nerve. It can be congenital or acquired.
Marcus Gunn Jaw Wink Phenomenon (Congenital Trigemino-Oculomotor Synkinesis)
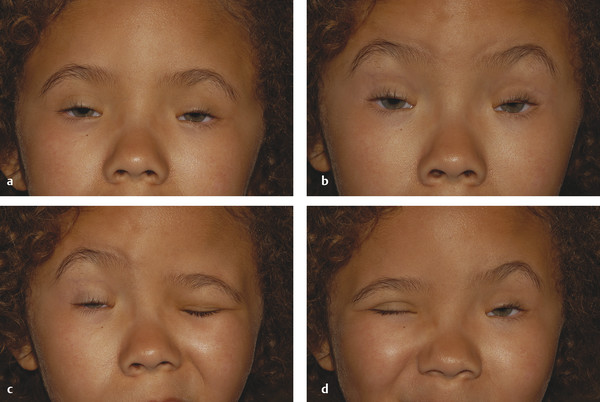
In this disorder, there is a central anomalous innervational pattern between the oculomotor and trigeminal nerves. The phenomenon is characterized by eyelid synkinesis with jaw movement. Characteristically, a unilateral ptosis of variable degree is noted shortly after birth. The ptotic eyelid is noted to open and close as the infant feeds. The phenomenon accounts for approximately 5% of congenital ptosis cases and may be associated with the following:
Amblyopia
Anisometropia
Strabismus
It may also be associated with a superior rectus palsy or a double elevator palsy (Fig. 7‑16).
There are two major groups of trigemino-oculomotor synkinesis:
External pterygoid–levator synkinesis in which the lid elevates when the jaw is thrust to the opposite side, when the jaw is projected forward, or when the mouth is widely opened.
Internal pterygoid–levator synkinesis in which the lid elevates on teeth clenching.
In some patients, the abnormal movements are only provoked by sucking. A rare condition in which the lid falls as the mouth opens has been referred to as the inverse Marcus Gunn jaw wink phenomenon.
It is generally believed that this condition improves with age, but this may be related to patients learning how to minimize the visible effects of the condition. Nevertheless, this should be borne in mind when discussing treatment options with patients and parents of affected children.
The treatment of this phenomenon is extremely difficult. It is important to ascertain whether the wink, the ptosis, or both are of concern to the patient or the parents. If the wink is mild and not of major concern, the ptosis can be treated according to the usual criteria applied to the management of ptosis (i.e., determined by the degree of levator function). If the wink is of concern, it can be treated either by an extirpation of the levator and a frontalis suspension procedure, performed unilaterally, or, more controversially, bilaterally or by means of a Lemagne procedure. The success of a Lemagne procedure relies on the patient having good preoperative levator function.
It can be very difficult to explain the pros and cons of this approach to the parents of a child with this condition. Naturally, most parents are reluctant to consent to surgery on the normal side. Although frontalis suspension surgery performed bilaterally can provide good symmetry in the absence of any complications of the surgery, it is important for the parents to appreciate that this will nevertheless result in an abnormal symmetry, with lag of both eyelids on downgaze. The extirpation of the levator muscle can be undertaken either via an anterior approach, with direct suturing of the sling material (e.g., autogenous fascia lata) to the tarsus, or via a posterior approach, with the sling material placed as a closed technique.
Aberrant Reinnervation of the Oculomotor Nerve
With aberrant reinnervation of the oculomotor nerve, there is an innervational anomaly within the neural sheath between the eyelid and other targets of the oculomotor nerve. It is characterized by inappropriate eyelid and extraocular muscle synkinesis. The disorder typically follows trauma or compression of the oculomotor nerve. The management of this disorder is particularly difficult. The aberrant eyelid movements can be halted by extirpating the levator muscle, which is followed by placement of a frontalis sling. The absence of a Bell’s phenomenon, however, places the patient at risk of exposure keratopathy. For this reason an initial trial of the use of a polypropylene suture is wise, because it is readily reversible.
Guillain–Barré Syndrome
Guillain–Barré syndrome, which may be generalized or may present as a bulbar variant, usually follows a febrile illness. Ptosis, which is usually of mild degree, is symmetrical and occurs in the context of a rapidly progressive bilateral ophthalmoplegia and facial diplegia. Classically, the cerebrospinal fluid (CSF) shows a raised protein level in the absence of a cellular response. A more limited form of Guillain–Barré syndrome, the Miller–Fisher variant, consists of bilateral ptosis and ophthalmoplegia associated with ataxia and areflexia but with no systemic weakness.
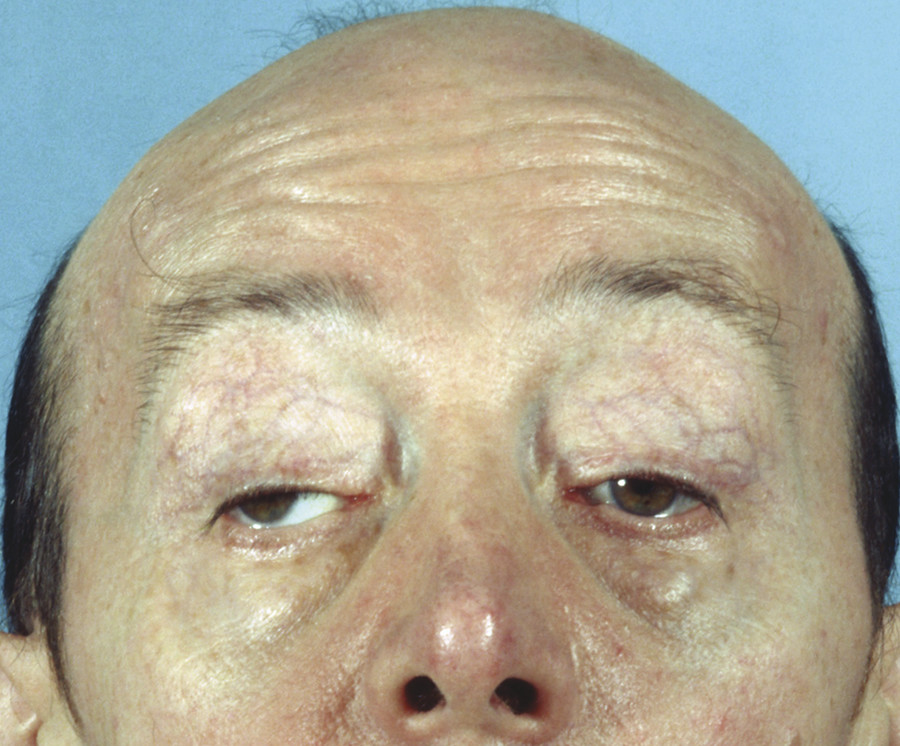
Cerebral Ptosis
A moderate to severe bilateral ptosis may occur after acute damage to the right cerebral hemisphere. The ptosis may be asymmetrical. A conjugate ocular deviation is also seen in this condition (Fig. 7‑17).
Botulism
Botulism, acquired through food poisoning, is a very rare neurological disorder characterized by ptosis and ophthalmoplegia, followed by dysarthria and dysphagia and then by weakness of the extremities.
Botulinum toxin blocks neuromuscular transmission and is commonly used therapeutically in the treatment of essential blepharospasm. It is being increasingly used by a variety of medical and nonmedical practitioners for the cosmetic improvement of periocular lines and wrinkles and to achieve a nonsurgical browlift. Blepharoptosis is a potential complication of the use of periocular botulinum toxin injections. The ptosis, which is usually partial and often unilateral when it occurs, is temporary and resolves completely after approximately 3 to 4 months as the effects of the botulinum toxin wear off. Patients undergoing periocular botulinum toxin injections should be warned about this risk. The ptosis may respond to the temporary topical administration of apraclonidine (Lopidine), an α-adrenergic receptor agonist as this stimulates Müller’s muscle, but the patient must be warned of the potential side effects of this glaucoma medication.
Key Point
A history of recent periocular botulinum toxin injections should always be sought in any patient presenting with an acquired ptosis.
Myogenic Ptosis
For a classification of myogenic ptosis, see Box 7.4.
Box 7.4 Myogenic Ptosis
Congenital “dystrophy” of the levator muscle
Myotonic dystrophy
Chronic progressive external ophthalmoplegia
Oculopharyngeal muscular dystrophy
Traumatic myopathy
Age-related levator muscle myopathy
Congenital “Dystrophy” of the Levator Palpebrae Superioris Muscle
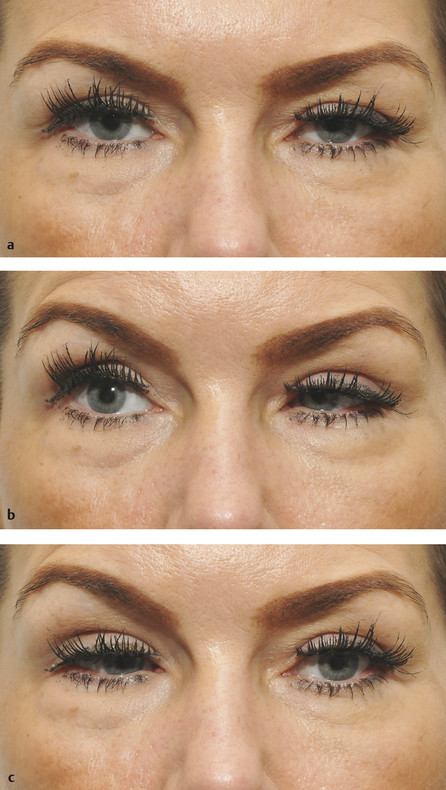
In congenital “dystrophy” of the levator palpebrae superioris muscle, the levator palpebrae superioris muscle is degenerate and replaced to a variable extent by fibrous and fatty tissue. The levator function varies from good to poor. The degree of ptosis can vary from minimal to severe and may interfere with visual development. Amblyopia associated with a congenital ptosis may be related to occlusion from the ptosis if it is severe enough to occlude the visual axis but may also be related to an undiagnosed refractive error or strabismus. The ptotic eyelid typically shows lag on downgaze (Fig. 7‑18).
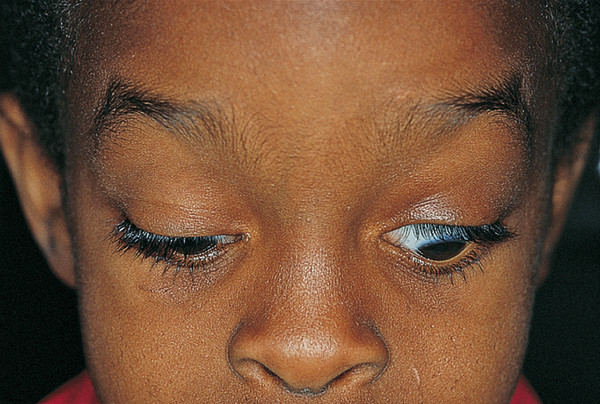
A congenital “dystrophic” ptosis may occur in isolation (simple congenital “dystrophic” ptosis), or it may be associated with a weakness of the superior rectus muscle (Fig. 7‑19). It may also be seen in the blepharophimosis–ptosis–epicanthus–inversus syndrome (BPES) or in congenital ocular fibrosis syndromes.
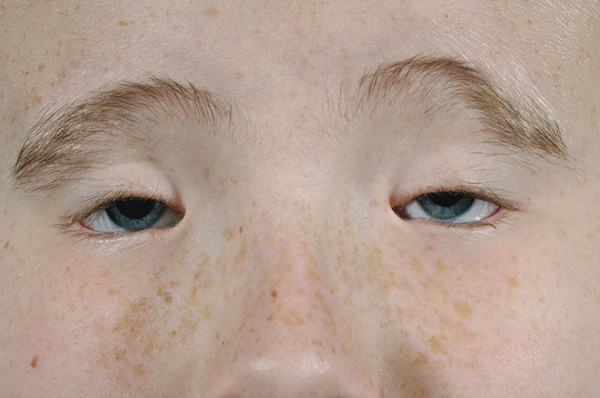
The BPES is an autosomal dominant disorder that comprises (Fig. 7‑20) several characteristics:
Bilateral ptosis, usually with poor levator function.
Blepharophimosis.
Telecanthus.
Epicanthus inversus (a fold of skin running upward and inward from the lower eyelid).
High arched eyebrows.
Two clinical subtypes have been described in which type 1, but not type 2, is associated with infertility related to primary ovarian failure. Patients may also have a lateral lower lid ectropion. There may also be a shortage of skin in the lateral aspect of the upper eyelids (Fig. 7‑20, Fig. 7‑21). Such patients often require a bilateral frontalis suspension procedure, usually after surgery to address the telecanthus and the epicanthus inversus.
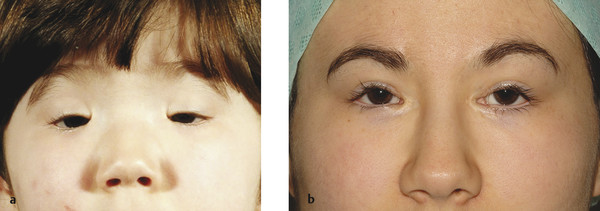
Patients with this disorder often have independent control of each eyebrow, which can pose particular cosmetic difficulties after a frontalis suspension procedure (Fig. 7‑22). This, and the position of the peak of the brows, needs to be taken into consideration before surgery.
The extraocular fibrosis syndromes are congenital ocular motility disorders that arise from dysfunction of the oculomotor, trochlear, and abducens nerves and/or the muscles that they innervate. Each is marked by a specific form of restrictive paralytic ophthalmoplegia with or without ptosis. Individuals with the classic form of congenital fibrosis of the extraocular muscles (CFEOM1) are born with bilateral ptosis and a restrictive infraductive external ophthalmoplegia. The absence of a Bell’s phenomenon leaves such patients at risk of exposure keratopathy from a frontalis suspension procedure.
Myotonic Dystrophy
Myotonic dystrophy is a rare autosomal dominant myopathic process that may be associated with a mild degree of symmetrical ptosis with a fair to poor degree of levator function (Fig. 7‑23). It is characterized by the following:
A progressive symmetrical external ophthalmoplegia.
Myotonia.
A myopathy with atrophy affecting the musculature of the face, neck, and limbs.
Classical posterior subcapsular cataracts.
The cataracts consist of small, colored crystalline opacities, or posterior, subcortical, and spoke-like opacities.
Classically, these patients demonstrate myotonia, a delayed relaxation after contraction, that is most noticeable on shaking hands when greeting the patient (grip myotonia). Myotonia can often be demonstrated by tapping a muscle (e.g., the thenar muscles) with a reflex hammer (percussion myotonia).
Men may show frontal balding and testicular atrophy.
Key Point
Patients with myotonic dystrophy typically have a poor Bell’s phenomenon and orbicularis oculi muscle weakness. They are at particular risk of exposure keratopathy after surgery. They are more likely to tolerate ptosis props than are other patients, because their orbicularis oculi muscle is weakened.
Other ocular signs associated with myotonic dystrophy include the following:
Pupillary light-near dissociation.
Ocular hypotonia.
Dry eyes.
A retinal pigmentary degeneration.
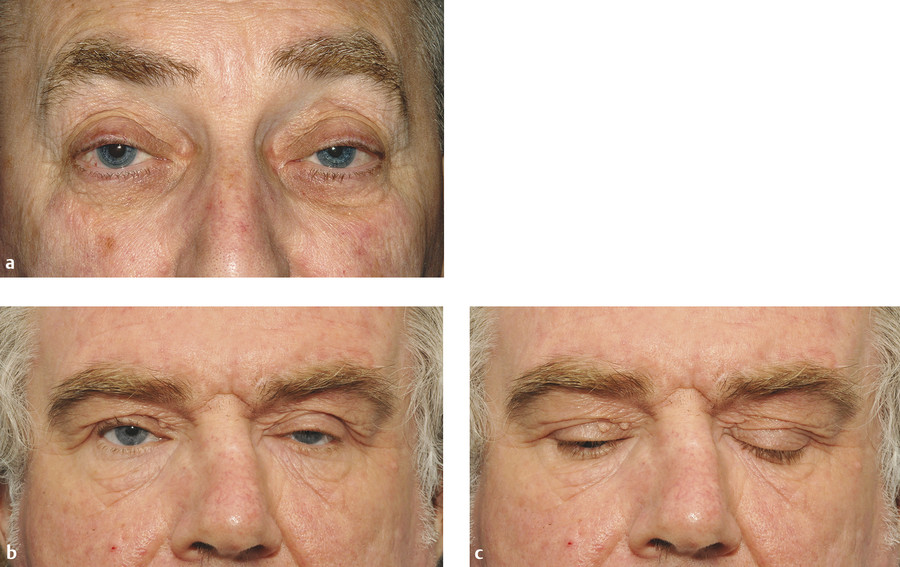
Chronic Progressive External Ophthalmoplegia
CPEO is a rare, slowly progressive disorder that is characterized by a progressive, symmetrical paralysis of the extraocular muscles, which do not respond to oculocephalic movements or to caloric stimulation. It may affect patients of all ages but typically manifests in the young adult years. Among the mitochondrial myopathies, CPEO is the most common manifestation, estimated at two-thirds of all incidences of mitochondrial-associated myopathies.
Clinical Features
Patients typically present with ptosis, which is often the presenting symptom (Fig. 7‑24). The first presenting symptom of ptosis is often unnoticed by the patient until the lids droop to the point of producing a visual field defect. Often patients will adopt an abnormal head posture with a chin elevation to compensate for the progressive ptosis. As the ptosis becomes more severe, the patient will show marked frontalis muscle overaction. The ptosis is typically bilateral but may be unilateral for a period of months to years before the fellow lid becomes involved.
Because the ophthalmoplegia is usually symmetrical, diplopia is a common complaint of these patients. In fact, the progressive ophthalmoplegia is often unnoticed until decreased ocular motility limits peripheral vision. Often, the ocular motility restriction is drawn to the patient’s attention by someone else. Patients tend to move their heads to adjust for the loss of peripheral vision caused by the horizontal ophthalmoplegia. Although all directions of gaze are affected, downgaze tends to be the least affected. Weakness of other muscle groups, including the orbicularis oculi, facial, and limb muscles, may be present in up to 25% of patients with CPEO. Frontalis muscle weakness may exacerbate the ptosis. Facial muscles may be involved, which leads to atrophy of facial muscle groups, producing a thin, expressionless face. Neck, shoulder, and extremity weakness with atrophy may affect some patients and can be mild or severe.
Mitochondrial deoxyribonucleic acid (DNA), which is maternally inherited, encodes proteins that are critical to the respiratory chain required to produce adenosine triphosphate (ATP). Deletions or mutations to segments of mitochondrial DNA lead to defective function of oxidative phosphorylation. This may be made evident in highly oxidative tissues like skeletal muscle and heart tissue. However, the extraocular muscles contain a volume of mitochondria that is several times greater than any other muscle group. As such, this results in the severe ocular symptoms of CPEO.
Diagnosis
The diagnosis of CPEO is confirmed by muscle biopsy. An examination of muscle fibers stained with Gomori trichrome stain reveals an accumulation of enlarged mitochondria. This produces a dark red staining of the muscle fibers, referred to as ragged red fibers. Although ragged red fibers are seen in normal aging, levels exceeding normal aging provide a diagnosis of a mitochondrial myopathy. In addition, performing polymerase chain reaction (PCR) testing, from a sample of a patient’s blood or muscle tissue, can determine a mutation of the mitochondrial DNA.
Management
There is currently no defined treatment to improve the muscle weakness of CPEO. Ptosis associated with CPEO may be managed by means of an anterior or posterior levator aponeurosis advancement procedure if the levator function is preserved, but a frontalis suspension procedure may be necessary. Surgery must be undertaken with great caution, however, because progressive weakness of the orbicularis oculi muscle, coupled with an absent Bell’s phenomenon, can place the patient at significant risk of exposure keratopathy.
Key Point
A levator aponeurosis advancement procedure for CPEO is best performed via a posterior eyelid approach. This affords a greater control of the final eyelid position and offers greater protection of the cornea in the early postoperative period, because the eyelid is initially low and the degree of postoperative lagophthalmos in the early postoperative period is less than that with an anterior approach.
Ophthalmoplegia Plus
The term ophthalmoplegia plus has been used to refer to a range of abnormalities that may be found with chronic progressive ophthalmoplegia. These abnormalities may be manifestations of associated neurodegenerative disorders. Kearns–Sayre’s syndrome (KSS) is a condition characterized by chronic progressive ophthalmoplegia, a retinal pigmentary degeneration, cardiac conduction defects often leading to complete heart block, and elevated CSF protein. It can affect young adults. It is important to perform dilated fundoscopy to determine whether there is a pigmentary retinopathy that may signify KSS. It is important to identify the cardiac conduction defect in such a patient by means of an electrocardiogram (ECG), because a cardiac pacemaker may be life-saving.
Oculopharyngeal Muscular Dystrophy
Oculopharyngeal muscular dystrophy is an autosomal dominant disorder that typically manifests in the sixth decade of life in patients with a mutation on the PABPN1 gene. Affected patients typically demonstrate a bilateral ptosis and a progressive external ophthalmoplegia. Dysphagia, facial weakness, and proximal limb weakness develop as the disorder progresses. A large number of such patients are of French-Canadian descent. The management of the ptosis is similar to that of ptosis-complicating CPEO (see above).
Muscle Trauma
A myogenic ptosis can occur after eyelid or orbital trauma (e.g., a “blow-in” fracture of the orbital roof). If the levator muscle has previously been transected after penetrating trauma, resulting in a complete ptosis with no levator function, it can be very difficult to differentiate a neurogenic ptosis from a myogenic ptosis.
Age-Related Levator Muscle/Müller’s Muscle Myopathy
Most age-related ptosis is due to a levator aponeurosis dehiscence but some older patients gradually develop a fatty degeneration of the levator muscle and/or Müller’s muscle with a sluggish levator function.
7.2.3 Aponeurotic Ptosis
For a classification of aponeurotic ptosis, see Box 7.5.
Aponeurotic ptosis is the result of a defect in the aponeurotic linkage between the levator muscle and the tarsal plate. It may be the result of a frank disinsertion of the aponeurosis from the tarsal plate or, much more commonly, a dehiscence or involutional stretching and redundancy of the aponeurosis.
Box 7.5 Aponeurotic Ptosis
Involutional (i.e., age-related)
Postsurgical (e.g., cataract surgery)
After eyelid trauma
After eyelid edema (e.g., blepharochalasis)
After long-term contact lens wear
Floppy eyelid syndrome
Chronic eyelid rubbing
Clinical Features
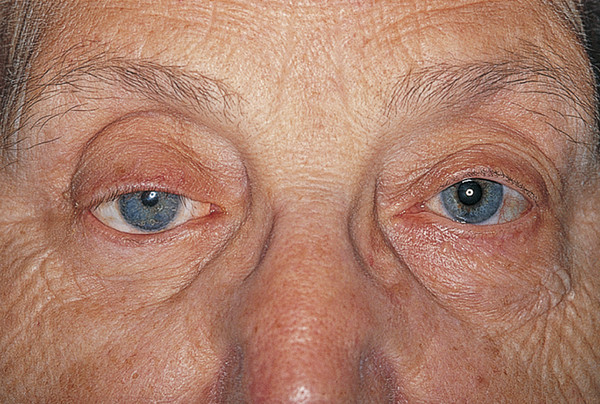
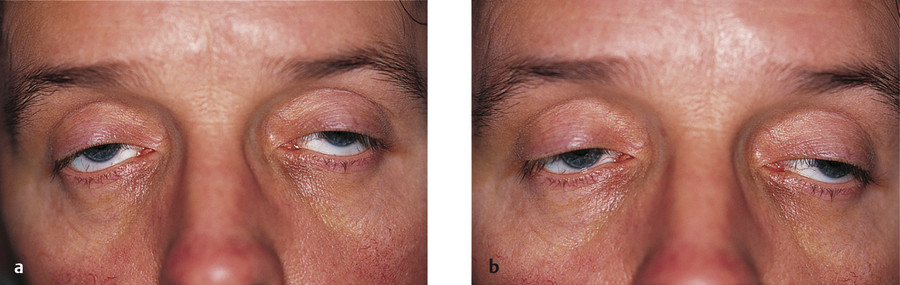
The following are the typical features of an aponeurotic ptosis (Fig. 7‑25):
A ptosis that is constant in all positions of gaze.
A lid drop on downgaze (Fig. 7‑25a).
Good levator function.
A high skin crease.
Thinning of the eyelid may occur, often with the iris visible through the closed eyelid (Fig. 7‑26). Patients with this visible iris sign, particularly those with atrophy of the preaponeurotic fat with a deep upper lid sulcus, are much more difficult to manage by means of an anterior-approach levator aponeurosis advancement. Such surgery can lead to a marked overcorrection of the lid position, contour abnormalities, and a visible scar. These patients are better managed with a posterior-approach Müller’s muscle resection or with a posterior-approach levator aponeurosis advancement, depending on the degree of ptosis.
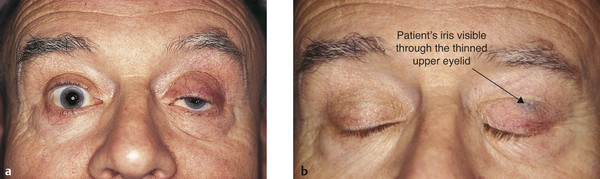
Patients typically notice worsening of the ptosis toward the end of the day, and this history of variability may lead to the suspicion of myasthenia.
Key Point
Particular care should be taken when making a skin crease incision in a patient with an aponeurotic ptosis, because the eyelid may be extremely thin. A corneal protector should be used. Patients with a visible iris sign are better managed with a posterior-approach Müller’s muscle resection or with a posterior-approach levator aponeurosis advancement.
7.2.4 Mechanical Ptosis
For a classification of mechanical ptosis, see Box 7.6.
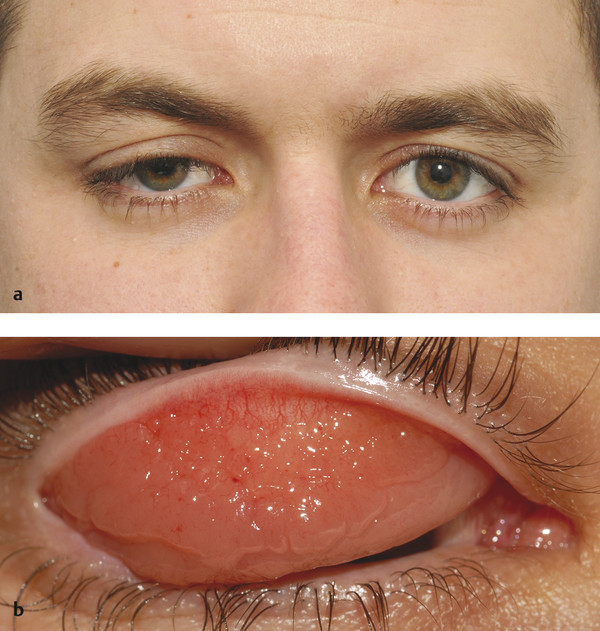
A variety of eyelid lesions can result in a secondary mechanical ptosis (Fig. 7‑27a,b), the management of which depends on the nature of the lesion (e.g., an upper lid capillary hemangioma may respond to the use of systemic Propranolol) (Fig. 7‑27c). Orbital lesions may present with a secondary mechanical ptosis (Fig. 7‑27d–i). Adhesions between the eyelid and the globe, such as in mucous membrane pemphigoid, may also result in a mechanical ptosis (Fig. 7‑27j,k).
Box 7.6 Mechanical Ptosis
Eyelid tumors
Orbital lesions
Cicatrizing conjunctival disorders
Foreign bodies
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


