Pathogenesis.
The pathogenetic mechanisms of ISD are unknown: in the vision that considers ISD as an autonomous disease, sebum metabolism and mycotic flora have been particularly investigated. The theories of excessive sebaceous gland activity from maternal hormones and nutritional factors such as biotin deficiency [23] have not been validated. Although the age of occurrence may suggest a role for maternal hormones, there is no evidence that this is the case. Other studies have demonstrated an abnormality with regard to essential fatty acids in the aetiology of ISD, but there has been no further corroboration [24]. Although there was no quantitative difference in sebum production between infants with ISD and unaffected control subjects, an altered fatty acid pattern has been demonstrated in ISD [25]. Some abnormal serum values have been identified in children with ISD during the active disease period. This has been explained by transiently impaired function of the enzyme Δ6-desaturase. On the basis of these laboratory findings, the role of (18-2)-linoleic acid and its metabolic products has been discussed in relation to the maintenance of the epidermal barrier function. In further studies, transepidermal water loss (TEWL), as a proxy for epidermal barrier function, was investigated on the forearms of 37 children with ISD and 25 healthy control subjects. Higher initial TEWL values normalized following local application of borage oil, rich in gamma-linoleic acid, the first metabolite of linoleic acid. Although a familial history of seborrhoea is quite often reported, there is no conclusive evidence that heredity has a role in ISD.
In regards to the mycotic hypothesis, most of the papers in this field delineate the importance of Malassezia furfur in the aetiology of the adult form of seborrheic dermatitis [26]. Its role in ISD has never been conclusively proven although this organism can activate complement in vitro by the alternative pathway and can trigger the ‘head and neck’ form of atopic dermatitis through a specific immunoglobulin E (IgE) response. In 1989, Broberg and Faergemann [27] demonstrated the presence of the organism in 18 out of 20 patients with ISD but in only 4 out of 20 control subjects. In the same year, Ruiz-Maldonado et al. [28] also detected M. furfur in 73% of patients with ISD, 33% of patients with atopic dermatitis or other dermatoses and in 53% of normal control subjects. In this paper the causative role of M. furfur was supported by the therapeutic benefit that followed treatment with topical ketoconazole. However, these studies are not conclusive and the current state of knowledge that the genus of Malassezia is (to date) composed of at least 13 species (one non-lipid-dependent species, i.e. M. pachydermatis, and 12 lipid-dependent species) [29], further reduces the significance of old investigations. Another previously held view was that ISD represents an ‘id’ reaction to Candida albicans skin and intestinal infection. In a study on a small cohort of children with ISD addressing this issue [21], Candida species were cultured from different sites in four out of the six patients who were studied, but no candidal agents could be demonstrated in either epidermis or dermis suggesting that Candida might not have a significant role in the pathogenesis of ISD.
Histopathology.
In contrast to adult seborrhoeic dermatitis, few histopathological investigations on ISD have been performed [21]. This is partially because of a common belief that the clinical diagnosis is straightforward (so a biopsy is not typically indicated) and also that the course of ISD is short and uncomplicated. In addition, the histology of the lesions is not entirely diagnostic and consists of a subacute dermatitis with concomitant features. However, Oranje et al. [21] studied six children and found a relatively homogeneous histological picture. Epidermal changes included parakeratosis (mostly patchy), microvesicles and a slight spongiosis, moderate acanthosis (psoriasiform in two patients) and a poorly formed granular layer. Dermal changes consisted of patchy mild perivascular infiltration of inflammatory lymphocytes and prominent perivascular oedema. Lymphocytes were often present within the epidermis. Direct immunofluorescence studies have not demonstrated significant deposits at either the perivascular or the epidermodermal junction in the dermis. A skin biopsy is mandatory when Langherhans cell histiocytosis (LCH) – a mimic of seborrhoeic dermatitis – is suspected. In LCH, the lesions are often more discrete and crusted and may present with more haemorrhagic lesions.
Clinical Features.
It is not surprising that the description of the clinical features of ISD can vary depending on the publication, and are even more accentuated in the spectrum of published clinical photographs from these reports.
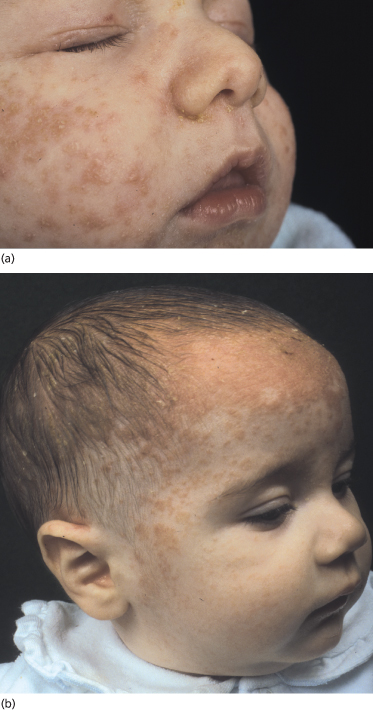
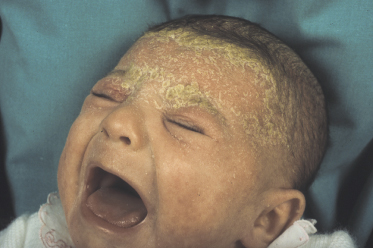
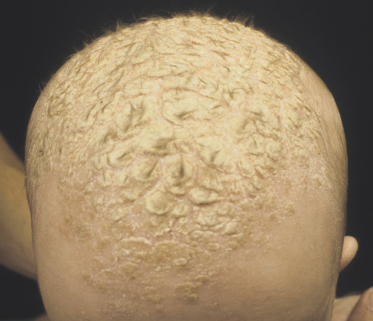
Classically, the disease begins in the first month of life and clears by 4–6 months of age (Fig. 35.2). In congenital cases, ISD manifests on the scalp as ‘cradle cap’, i.e. yellowish or whitish, greasy, thick, adherent, frequently confluent scales, mostly occurring on the vertex and frontal areas. In the acquired forms, ISD may appear on the previously clear scalp areas of affected infants (Figs 35.3 and 35.4) [30]. The eruption also appears concurrently on the face and in the flexural areas. On the face, small round or oval erythrosquamous areas are seen, in particular on the forehead, eyebrows, retro-auricular area, nasolabial folds and the sides. The lesions tend easily to coalesce. Quite commonly, lesions develop in the neck, axillae, umbilicus, inguinal and intergluteal folds (the so-called: ‘bipolar’ ISD) (Fig. 35.5). The rash starts on the scalp and shows relatively little inflammation, but in intertriginous areas scales are much less pronounced than those on the scalp and become rapidly macerated and eroded, so that desquamation can be noted only at the periphery of the lesions.
Fig. 35.2 (a) Initial infantile seborrhoeic dermatitis (ISD) affecting exclusively the face in a mild form. (b) The yellowish-orange aspect is very suggestive of ISD.
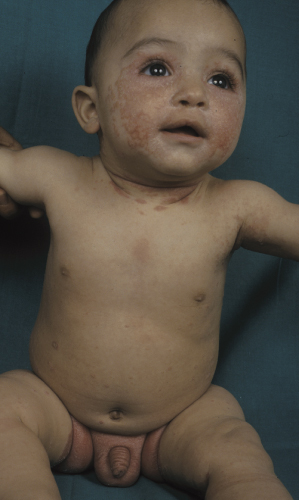
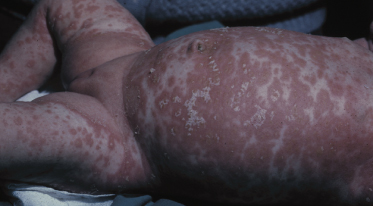
In the napkin area ISD can be largely influenced by many different factors including type of napkin, frequency of changes, hygienic habits and bowel movements, so that individual lesions may coalesce into large erythematous infiltrated plaques (Fig. 35.6) [31,32]. Only exceptionally, lesions may appear on the chest or the baby becomes erythrodermic (Fig. 35.7) [33]. Leiner’s disease has at times been used to describe the erythrodermic stage of ISD [34].

Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


