It is widely accepted that up to 25% of all persons experience at least one episode of urticaria during their lifetime and that infections represent the most frequent underlying cause in childhood. Moreover, urticaria and/or angioedema may be the presenting signs in more than 80% of paediatric patients suffering from anaphylaxis [14,15]. However, the exact incidence of drug-related urticaria, which is mostly of the acute type in children and adolescents, remains to be determined. Nevertheless, urticaria is regarded as the second most common drug eruption, accounting for an estimated 15–20% of all cutaneous drug reactions [16].
Pathophysiologically, drug-induced urticaria most often corresponds to an immediate (type I) hypersensitivity reaction. In sensitized patients, mast cells (MC) carry drug-specific IgE antibodies (sIgE) bound to the high-affinity IgE receptor (IgεRI) on their surface. Usually within minutes to 2 hours after oral intake, the culprit drug cross-links sIgE molecules, thus inducing MC mediator release. In childhood, drugs most frequently involved in immediate-type drug allergy are beta-lactam antibiotics (penicillin, amoxicillin, cephalosporins), sulphonamides and myorelaxants [17,18].
Intriguingly, acute drug-induced urticaria may also be due to non-immunological intolerance reactions. Most importantly, non-steroidal anti-inflammatory drugs (NSAIDs), a pharmacologically heterogeneous group of cyclo-oxygenase inhibitors, are known to skew arachidonic acid metabolism towards the 5-lipoxygenase pathway, thus enhancing the synthesis of proinflammatory cysteinyl leukotrienes. As a clinical consequence, 0.5–4% of children treated with NSAIDs such as ibuprofen, diclofenac or paracetamol (acetominophen) may develop acute urticaria within minutes to a maximum of 24 hours after drug ingestion. Concurrent facial angioedema manifests in an age-dependent manner: in fewer than 5% of infants and toddlers, but in up to 20% of adolescents and young adults with NSAID intolerance, acute urticaria is accompanied by facial oedema. It is also noteworthy that more than 80% of intolerant children will cross-react upon challenge with another NSAID [19–22].
Besides discontinuing exposure to the suspected trigger, systemic antihistamines represent the mainstay of treatment of acute drug-induced urticaria. Second-generation H1-antagonists such as cetirizine, loratadine or ketotifen should be preferentially used in order to avoid ADRs that are frequently associated with first-generation antihistamines, particularly drowsiness. In contrast, it is still a matter of debate whether so-called third-generation H1-antihistamines such as desloratadine, levocetirizine or fexofenadine are more efficacious in the therapy of acute urticaria than their predecessors. Non-responsive patients may benefit from short-term systemic glucocorticoid therapy, for example methylprednisolone 1 mg/kg/day over 3 days, although the efficacy of this treatment modality remains to be verified in controlled clinical trials [13,23].
Serum Sickness-Like Reactions
Serum sickness-like reactions (SSLRs) may occur during the first course of treatment with the culprit agent and usually arise within 1 to 3 weeks after drug initiation. Typical symptoms comprise low-grade fever, arthralgia and an urticarial, sometimes morbilliform, scarlatiniform or polymorphous rash that is often only mildly pruritic and self-limited. Lymphadenopathy and eosinophilia may also be present; however, in contrast to true serum sickness, characteristic laboratory changes, vasculitis or renal involvement are not regularly encountered [24].
Epidemiological data on SSLRs are still very scarce, but this hypersensitivity reaction is known to mostly affect infants and children. Accordingly, the estimated pooled incidence of cefaclor-related SSLRs has been calculated in the range 0.02–0.2% per drug course in paediatric patients. Moreover, epidemiological investigations suggest that the risk of SSLR is greater under treatment with cefaclor than with any other antibiotic therapy, including further cephalosporins. A retrospective cohort study disclosed that the relative risk of SSLRs for cefaclor compared with amoxicillin was 19 : 1. Furthermore, the WHO database for international drug monitoring registered 722 reports of SSLRs to cefaclor compared with only 34 for amoxicillin and 12 for cephalexin [25,26]. Hence, even if prospective controlled trials are lacking, cefaclor can be regarded as the principal eliciting drug in SSLRs. This is also corroborated by a considerable number of case reports and case series published during the last two decades [27–30].
Other drugs that have been implicated in causing SSLRs include biological agents (efalizumab [31], omalizumab [32], rituximab [33], infliximab [34]), antibiotics (cefuroxim [35], cefazolin [36], meropenem [37], minocycline [38], ciprofloxacin [39], rifampicin [40]), antimycotics (griseofulvin [41], itraconazole [42]) and other agents such as bupropion [43], clopidogrel [44], fluoxetine [45], N-acetylcysteine [46] or streptokinase [47] .
In vitro parameters that are characteristically altered in true type III serum sickness such as immune complexes, hypocomplementaemia or albuminuria are not regularly observed in SSLRs. This clearly implies that serum sickness reactions and SSLRs are clinically similar disorders, but arise due to distinct hypersensitivity mechanisms. More than 15 years ago, Kearns and co-workers postulated that a reactive cefaclor metabolite may be generated in genetically susceptible hosts, and bind with tissue proteins to elicit an inflammatory response manifesting as SSLR. However, this pathogenetic model still awaits clinical and experimental corroboration [48].
As the underlying cause of SSLRs remains unknown, its treatment is purely symptomatic, mainly consisting of discontinuation of the offending agent, antihistamines in case of urticaria, and NSAIDs for patients with arthralgia and/or arthritis. As in drug-induced urticaria, it is still unclear whether a short course of systemic glucocorticoids is a viable treatment option in SSLR patients with persisting symptoms despite antihistamine therapy. However, in a retrospective study of 31 children presenting to the emergency department for cefaclor-induced SSLR, a combination of oral prednisone and an antihistamine was found to be the preferred medication prescribed by the majority of treating paediatricians [49].
References
1 WHO. International drug monitoring: the role of national centres. Report of a WHO meeting. World Health Org Tech Rep Ser 1972;498:1–25.
2 Rawlins MD, Thompson JW. Pathogenesis of adverse drug reactions. In: Davies DM (ed.) Textbook of Adverse Drug Reactions. Oxford: Oxford University Press, 1977; 10.
3 Johansson SG, Bieber T, Dahl R et al. Revised nomenclature for allergy for global use: Report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol 2004;113:832–6.
4 Pichler WJ, Adam J, Daubner B, Gentinetta T, Keller M, Yerly D. Drug hypersensitivity reactions: pathomechanism and clinical symptoms. Med Clin North Am 2010;94:645–64, xv.
5 Impicciatore P, Choonara I, Clarkson A, Provasi D, Pandolfini C, Bonati M. Incidence of adverse drug reactions in paediatric in/out-patients: a systematic review and meta-analysis of prospective studies. Br J Clin Pharmacol 2001;52:77–83.
6 Menniti-Ippolito G, Raschetti R, Da CR, Giaquinto C, Cantarutti L. Active monitoring of adverse drug reactions in children. Italian Paediatric Pharmacosurveillance Multicenter Group. Lancet 2000;355:1613–14.
7 Ibia EO, Schwartz RH, Wiedermann BL. Antibiotic rashes in children: a survey in a private practice setting, Arch Dermatol 2000;136:849–54.
8 Bourgeois FT, Mandl KD, Valim C, Shannon MW. Pediatric adverse drug events in the outpatient setting: an 11-year national analysis. Pediatrics 2009;124:e744–e750.
9 Clavenna A, Bonati M. Adverse drug reactions in childhood: a review of prospective studies and safety alerts, Arch Dis Child 2009;94:724–8.
10 Rebelo GE, Fonseca J, Araujo L, Demoly P. Drug allergy claims in children: from self-reporting to confirmed diagnosis. Clin Exp Allergy 2008;38:191–8.
11 Lange L, Koningsbruggen SV, Rietschel E. Questionnaire-based survey of lifetime-prevalence and character of allergic drug reactions in German children. Pediatr Allergy Immunol 2008;19:634–8.
12 Khan DA, Solensky R. Drug allergy. J Allergy Clin Immunol 2010;125:S126–S137.
13 Zuberbier T, Asero R, Bindslev-Jensen C et al. EAACI/GA(2)LEN/EDF/WAO guideline: definition, classification and diagnosis of urticaria, Allergy 2009;64:1417–26.
14 Mehl A, Wahn U, Niggemann B. Anaphylactic reactions in children – a questionnaire-based survey in Germany. Allergy 2005;60:1440–5.
15 de Silva IL, Mehr SS, Tey D, Tang ML. Paediatric anaphylaxis: a 5 year retrospective review, Allergy 2008;63:1071–6.
16 Tan EK, Grattan CE. Drug-induced urticaria. Expert Opin Drug Saf 2004;3:471–84.
17 Shin HT, Chang MW. Drug eruptions in children. Curr Probl Pediatr 2001;31:207–34.
18 Segal AR, Doherty KM, Leggott J, Zlotoff B. Cutaneous reactions to drugs in children. Pediatrics 2007;120:e1082–e1096.
19 Sanchez-Borges M. NSAID hypersensitivity (respiratory, cutaneous, and generalized anaphylactic symptoms). Med Clin North Am 2010;94:853–64, xiii.
20 Sanchez-Borges M, Capriles-Behrens E, Caballero-Fonseca F. Hypersensitivity to non-steroidal anti-inflammatory drugs in childhood. Pediatr Allergy Immunol 2004;15:376–80.
21 Kidon MI, Kang LW, Chin CW, Hoon LS, Hugo VB. Nonsteroidal anti-inflammatory drug hypersensitivity in preschool children. Allergy Asthma Clin Immunol 2007;3:114–22.
22 Kidon MI, Kang LW, Chin CW et al. Early presentation with angioedema and urticaria in cross-reactive hypersensitivity to nonsteroidal antiinflammatory drugs among young, Asian, atopic children. Pediatrics 2005;116:e675–e680.
23 Del CA, Sastre J, Montoro J et al. Use of antihistamines in pediatrics, J Investig Allergol Clin Immunol 2007;17(Suppl. 2):28–40.
24 Shah KN, Honig PJ, Yan AC. “Urticaria multiforme”: a case series and review of acute annular urticarial hypersensitivity syndromes in children, Pediatrics 2007;119:e1177–e1183.
25 Heckbert SR, Stryker WS, Coltin KL, Manson JE, Platt R. Serum sickness in children after antibiotic exposure: estimates of occurrence and morbidity in a health maintenance organization population. Am J Epidemiol 1990;132:336–42.
26 Stricker BH, Tijssen JG. Serum sickness-like reactions to cefaclor. J Clin Epidemiol 1992;45:1177–84.
27 Sanklecha MU. Cefaclor induced serum sickness like reaction. Indian J Pediatr 2002;69:921.
28 Isaacs D. Serum sickness-like reaction to cefaclor. J Paediatr Child Health 2001;37:298–9.
29 Parra FM, Igea JM, Martin JA, Alonso MD, Lezaun A, Sainz T. Serum sickness-like syndrome associated with cefaclor therapy. Allergy 1992;47:439–40.
30 Hebert AA, Sigman ES, Levy ML. Serum sickness-like reactions from cefaclor in children. J Am Acad Dermatol 1991;25:805–8.
31 Shraf-Benson S, Wall GC, Veach LA. Serum sickness-like reaction associated with efalizumab. Ann Pharmacother 2009;43:383–6.
32 Pilette C, Coppens N, Houssiau FA, Rodenstein DO. Severe serum sickness-like syndrome after omalizumab therapy for asthma. J Allergy Clin Immunol 2007;120:972–3.
33 Finger E, Scheinberg M. Development of serum sickness-like symptoms after rituximab infusion in two patients with severe hypergammaglobulinemia. J Clin Rheumatol 2007;13:94–5.
34 Gamarra RM, McGraw SD, Drelichman VS, Maas LC. Serum sickness-like reactions in patients receiving intravenous infliximab. J Emerg Med 2006;30:41–4.
35 Baniasadi S, Fahimi F, Mansouri D. Serum sickness-like reaction associated with cefuroxime and ceftriaxone. Ann Pharmacother 2007;41:1318–19.
36 Brucculeri M, Charlton M, Serur D. Serum sickness-like reaction associated with cefazolin. BMC Clin Pharmacol 2006;6:3.
37 Ralph ED, John M, Rieder MJ, Bombassaro AM. Serum sickness-like reaction possibly associated with meropenem use. Clin Infect Dis 2003;36:E149–E151.
38 Landau M, Shachar E, Brenner S. Minocycline-induced serum sickness-like reaction. J Eur Acad Dermatol Venereol 2000;14:67–8.
39 Guharoy SR. Serum sickness secondary to ciprofloxacin use. Vet Hum Toxicol 1994;36:540–1.
40 Parra FM, Perez Elias MJ, Cuevas M, Ferreira A. Serum sickness-like illness associated with rifampicin. Ann Allergy 1994;73:123–5.
41 Colton RL, Amir J, Mimouni M, Zeharia A. Serum sickness-like reaction associated with griseofulvin. Ann Pharmacother 2004;38:609–11.
42 Park H, Knowles S, Shear NH. Serum sickness-like reaction to itraconazole. Ann Pharmacother 1998;32:1249.
43 Waibel KH, Katial RK. Serum sickness-like reaction and bupropion. J Am Acad Child Adolesc Psychiatry 2004;43:509.
44 Phillips EJ, Knowles SR, Shear NH. Serum sickness-like reaction associated with clopidogrel. Br J Clin Pharmacol 2003;56:583.
45 Shapiro LE, Knowles SR, Shear NH. Fluoxetine-induced serum sickness-like reaction. Ann Pharmacother 1997;31:927.
46 Mohammed S, Jamal AZ, Robison LR. Serum sickness-like illness associated with N-acetylcysteine therapy. Ann Pharmacother 1994;28:285.
47 Schweitzer DH, van der Wall EE, Bosker HA, Scheffer E, Macfarlane JD. Serum-sickness-like illness as a complication after streptokinase therapy for acute myocardial infarction. Cardiology 1991;78:68–71.
48 Kearns GL, Wheeler JG, Childress SH, Letzig LG. Serum sickness-like reactions to cefaclor: role of hepatic metabolism and individual susceptibility. J Pediatr 1994;125:805–11.
49 Joubert GI, Hadad K, Matsui D, Gloor J, Rieder MJ. Selection of treatment of cefaclor-associated urticarial, serum sickness-like reactions and erythema multiforme by emergency pediatricians: lack of a uniform standard of care, Can J Clin Pharmacol 1999;6:197–201.
Exanthematous Drug Eruptions
Maculopapular Exanthems
Maculopapular exanthems (MPEs) represent the most common adverse drug reactions affecting the skin. Historical and more recent cohort studies have shown that they account for 35–90% of all cutaneous ADRs, depending on the investigated populations of varying age, with different underlying diseases in variable clinical settings [1–4].
Literally all xenobiotic therapeutic agents may cause MPE, and the majority of licensed drugs induce exanthematous rashes in 1% or more of treated patients. In children, beta-lactam antibiotics (penicillins, cefaclor, other cephalosporins, sulfamethoxazole) and aromatic anticonvulsants (phenytoin, phenobarbital, carbamazepine) most frequently elicit MPE, whereas other culprit drugs such as macrolide antibiotics, thiazide diuretics, NSAIDs or benzodiazepines are less frequently encountered [5].
Although its complex pathophysiology remains to be fully elucidated, drug-induced MPE is generally considered to be a non-immediate, T-cell-mediated hypersensitivity reaction of type IVc according to the modified Gell and Coombs classification. Briefly, highly activated, drug-specific CD4+ and CD8+ T-cells infiltrate the cutis accompanied by eosinophils and macrophages as well as different dendritic cell subpopulations. Allergen-specific T-cells synthesize and liberate cytotoxic granule proteins such as perforin and granzyme B, thus causing keratinocyte damage, which manifests as vacuolar damage upon histology. Moreover, T-cells and other leucocytes as well as resident skin cells release a heterogeneous profile of cytokines and chemokines that, terminally, enhance inflammatory processes and contribute to the recruitment of eosinophils, which are often, but not always, detectable upon lesional skin biopsy [5,6].
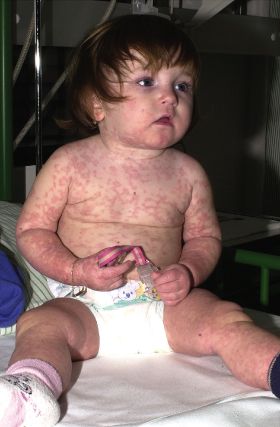
Maculopapular exanthem typically occurs within the second week after start of treatment, but may also first appear one or two days after the end of drug therapy (‘eruption of the ninth day’). In contrast, sensitized individuals may reveal a faster disease onset, developing skin symptoms within the first day of drug re-exposure. Characteristic cutaneous features consist of symmetrically distributed, erythematous, sometimes salmon-coloured macules and papules that usually coalesce into a morbilliform (Fig. 183.2), scarlatiniform or rubelliform rash. However, MPEs are clinically heterogeneous, sometimes manifesting as annular, urticarial, purpuric or serpiginous eruptions (Fig. 183.3). Therefore the differential diagnosis of drug-induced exanthemas is broad including, among others, viral infections (Epstein–Barr virus, parvovirus B19, human herpesvirus (HHV) 6, adenovirus, etc.), bacterial infections (Streptococcus pyogenes, Treponema pallidum, etc.), systemic juvenile arthritis (Still disease), Kawasaki syndrome, graft-versus-host-disease (GvHD) and erythema multiforme. In general, MPEs spare the palmoplantar region and mucous membranes, are only moderately pruritic and usually not associated with high-grade fever or general malaise [7].
Fig. 183.2 Morbilliform maculopapular exanthem occurring after five days of treatment with amoxicillin.
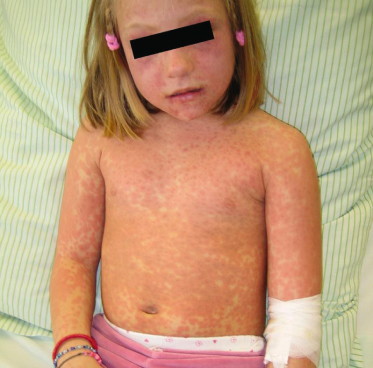
Fig. 183.3 Atypical, erythema infectiosum-like drug rash first observed after four days of oral amoxicillin treatment.
Uncomplicated exanthematous rashes resolve spontaneously without sequelae within a few days after drug withdrawal. Hence, systemic glucocorticoid treatment (e.g. methylprednisolone 1 mg/kg/day over 3–5 days) can usually be reserved for children with marked pruritus. Yet, blister formation, evolving erythroderma, mucous membrane involvement, atypical target lesions and the involvement of other organ systems have to be regarded as signs of impending serious drug rashes that should prompt immediate hospital admission for further diagnostic workup and intensified treatment under close medical supervision (see below).
DRESS Syndrome: Drug Rash with Eosinophilia and Systemic Symptoms
DRESS syndrome, also called drug-induced or anticonvulsant hypersensitivity syndrome (DIHS/AHS), represents a rare but acute and potentially fatal multisystem drug reaction. Basically, it is characterized by the clinical tetrad of fever, maculopapular rash, lymphadenopathy and internal organ involvement. Most strikingly, the onset of DRESS may be delayed for more than 3 weeks after initial drug exposure whereas symptoms regularly persist for longer than 2 weeks after discontinuation of the causative agent [8].
The exact incidence of DRESS is unknown, but it is thought to occur in approximately 1 : 1000 to 1 : 10,000 of exposures to aromatic anticonvulsants [9]. In children and adolescents, the drugs most frequently implicated are the aromatic anticonvulsants (phenytoin, carbamazepine, phenobarbital, oxcarbazepine) and lamotrigine [10,11], but other agents have also been described as causes of paediatric DRESS, i.e. allopurinol [12], vancomycin [13], minocycline [13], sulfasalazine [14], aspirin [15], nevirapine [16] and dapsone [17]. Interestingly, amoxicillin has been shown to elicit flare-ups in adolescents with DRESS syndrome previously induced by other drugs [18].
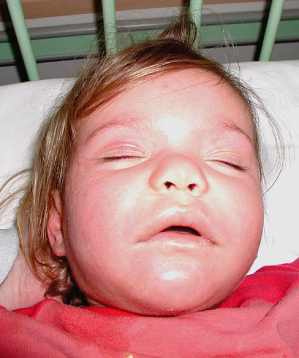
The first non-specific clinical signs are a fever in excess of 38.5°C and malaise, which may precede cutaneous and internal symptoms by several days. Approximately 85% of all patients then develop a rash; this has recently been shown to occur in up to 100% of paediatric patients with anticonvulsant-induced DRESS syndrome [10,19]. Cutaneous eruptions may be polymorphous and vary during the clinical course. Initially, starting in the facial region and spreading craniocaudally, non-specific maculopapular and often morbilliform rashes are observed, which later become infiltrated showing a follicular accentuation [20,21]. In children, cutaneous symptoms may evolve into erythroderma, vesiculobullous lesions, targetoid or even blistering eruptions in 44%, 16%, 13% and 9% of patients, respectively. While mucosal lesions such as cheilitis, conjunctivitis and pharyngitis appear in about 50% of children and are usually discrete, up to 70% of children reveal massive facial oedema (Fig. 183.4), sometimes with a characteristic periorbital accentuation. Nearly 90% of patients will develop some degree of lymphadenopathy at a minimum of two sites, with enlarged (>2 cm) lymph nodes particularly in the cervical region, but general lymphadenopathy can also be observed [10].
Fig. 183.4 Marked facial oedema and malaise in a child with DRESS syndrome.
Courtesy of Dr Lars Lange, Department of Pediatric Allergology and Pulmonology, Marienhospital, Bonn, Germany.
In a sizeable number of patients, internal organ involvement may be delayed by 1–4 weeks after the onset of skin symptoms. Whereas some individuals suffer from only mild systemic symptoms, others may be afflicted with severe multiorgan failure accounting for a mortality of 10% in adult patients with DRESS syndrome [22]. Most commonly observed, hepatic dysfunction may range from an isolated elevation of serum liver enzymes to acute hepatitis and hepatic necrosis with terminal liver failure [23–25]. Acute kidney injury is the second most commonly observed internal manifestation of DRESS syndrome. Affected patients reveal isolated haematuria or interstitial nephritis, but may also develop acute renal failure requiring transient dialysis [26–28]. Other less common, but clinically relevant systemic reactions involve the central nervous system (encephalitis, aseptic meningitis), the bronchopulmonary system (pneumonitis, respiratory distress syndrome), myocarditis, transient thyroiditis, colitis and pancreatitis [20].
Haematological abnormalities include peripheral leucocytosis (>11 × 109/L), atypical lymphocytosis (>5%) and eosinophilia (>1.5 × 109/L) occurring in more than 70% of patients within the first 2–3 weeks of DRESS manifestation. Further non-specific changes detected in peripheral blood are neutropenia, agranulocytosis and thrombocytopenia [8].
The pathogenesis of DRESS syndrome has not been fully elucidated. However, the currently most widely accepted pathogenetic model postulates that, in a first step, genetically predisposed individuals metabolize the culprit drug into a reactive compound that cannot be detoxified due to genetically determined abnormalities within the required enzyme systems. As a consequence, the bioactivated drug metabolite irreversibly modifies cellular proteins, which then serve as targets for an immune-mediated type IVb hypersensitivity reaction orchestrated by activated T-cells secreting large amounts of interleukin-5 and interferon-γ. Finally, this massive drug-induced immune stimulation may lead to reactivation of latent lymphotropic viruses, particularly HHV-6, which could be responsible for the markedly chronic course in patients with DRESS syndrome [6].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


