+, Occasional finding; ++ common finding.
BCC, basal cell carcinoma; CALM, café-au-lait macules; CMC, chronic mucocutaneous candidiasis; DLE, discoid lesions of lupus erythematosus; GVHD, graft-versus-host disease; IL, interleukin; IPEX, ; IRAK4, IL-1 receptor associated kinase-4; JIA, juvenile idiopathic arthritis; SVV, small vessel vasculitis; LE, lupus erythematosus; PG, pyoderma gangrenosum; SCID, severe combined immunodeficiency; TAP, transporter associated with processing; WHIM, warts, hypogammaglobulinaemia, infections, and myelokathexis.
* Extensive cutaneous and extracutaneous granulomatous disease has been described in children with hypomorphic RAG1 or RAG2 mutations.
† Patients with the autosomal recessive form of hyper-IgE syndrome are also at increased risk of developing mucocutaneous squamous cell carcinomas and severe warts, molluscum contagiosum and herpes simplex or varicella-zoster viral infections..
In general, immunodeficiency should be suspected when patients have recurrent infections of increased duration and severity or caused by unusual organisms [7]. Incomplete clearance of infections or a poor response to antibiotic therapy may represent additional signs. Other frequent extracutaneous abnormalities in children with immunodeficiencies include failure to thrive, chronic diarrhoea, hepatosplenomegaly, lymphadenopathy or lack of expected nodes, and haematological abnormalities. Malignancy is the second leading cause of death (after infection) in children and adults with congenital immunodeficiency disorders [8].
References
1 Geha RS, Notarangelo LD, Casanova JL et al. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J Allergy Clin Immunol 2007;120:776–94.
2 Buckley RH. Primary immunodeficiency diseases due to defects in lymphocytes. N Engl J Med 2000;343:1313–24.
3 Fischer A. Human primary immunodeficiency diseases. Immunity 2007;27:835–45.
4 Maródi L, Notarangelo LD. Immunological and genetic bases of new primary immunodeficiencies. Nat Rev Immunol 2007;7:851–61.
5 Mitra A, Pollock B, Gooi J. Cutaneous granulomas associated with primary immunodeficiency disorders. Br J Dermatol 2005;153:194–9.
6 Torrelo A, Vera A, Portugues M. Perforating neutrophilic and granulomatous dermatitis of the newborn: a clue to immunodeficiency. Pediatr Dermatol 2007;24:211–5.
7 Slatter MA, Gennery AR. An approach to the patient with recurrent infections in childhood. Clin Exp Immunol 2008;152:389–96.
8 Mueller BU, Pizzo PA. Cancer in children with primary or secondary immunodeficiencies. J Pediatr 1995;126:1–10.
Ataxia Telangiectasia
Ataxia telangiectasia (AT) is an autosomal recessive disorder characterized by cerebellar ataxia beginning in infancy, progressive oculocutaneous telangiectasias, recurrent sinopulmonary infections and chromosomal instability with hypersensitivity to ionizing radiation. The incidence of AT is estimated to be 1 in 50,000 to 1 in 100,000 births.
Pathogenesis.
Ataxia telangiectasia is caused by loss-of-function mutations in both copies of the ataxia-telangiectasia mutated (ATM) gene, which encodes a serine/threonine protein kinase that resembles phosphatidylinositol 3-kinases [1,2]. The MRE11-RAD50-NBS1 complex helps to recruit and activate the ATM protein in response to DNA damage, especially double-stranded breaks [3]. ATM subsequently phosphorylates (and thereby activates) multiple targets such as p53, BRCA1, FANCD2, Artemis, MRE11 and NBS1, which leads to cell cycle arrest at G1/S and facilitates DNA repair or, in the event of severe DNA damage, apoptosis. DNA breaks occur not only upon exposure to ionizing radiation, but also during physiological processes such as V(D)J recombination in lymphocytes, telomere maintenance and meiosis, therefore explaining the sensitivity to ionizing radiation, immunodeficiency, premature ageing and defective spermatogenesis that are observed in patients with AT. The progressive neurological deterioration of AT is thought to result from defective DNA repair in this population of cells that are unable to replicate.
Clinical Features.
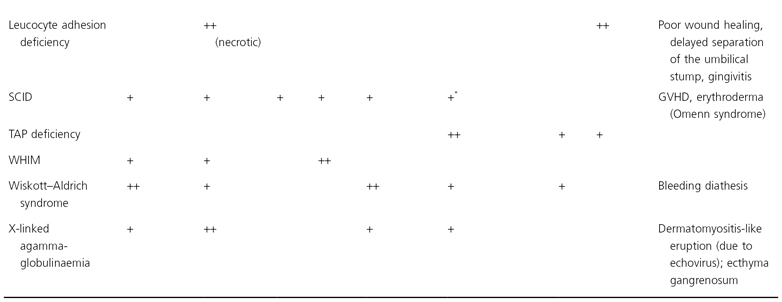
The initial manifestation is usually ataxia, which typically becomes apparent when the child begins to walk. However, diagnosis of AT is often delayed until telangiectasias are noted, usually between 3 and 6 years of age. The telangiectasias first appear on the bulbar conjunctivae at the corners of the eyes and extend toward the iris as red symmetrical horizontal streaks (Fig. 177.1). During the next few years, patients begin to develop cutaneous telangiectasias, especially on the ears, eyelids, malar prominences, V of the neck and antecubital and popliteal fossae. Less commonly, telangiectasias are observed on the dorsal aspects of the hands and feet and the palate. Although the ocular telangiectasias are striking, the cutaneous telangiectasias may be subtle and resemble fine petechiae, especially in extrafacial sites.
Fig. 177.1 Telangiectasias of the bulbar conjuctiva in ataxia telangiectasia are particularly prominent at the canthi.
Evidence of premature ageing of the skin and hair is noted in almost 90% of affected individuals [4]. Subcutaneous fat is lost during childhood and facial skin tends to become atrophic and sclerotic. Grey hairs may be found in young children, and diffuse greying of the hair often occurs by adolescence. Non-infectious cutaneous granulomas (Fig. 177.2) frequently develop and have a tendency to ulcerate [5]. Other skin findings associated with AT are large café-au-lait spots and other forms of ‘pigmentary mosaicism’ (presumably reflecting chromosomal instability) [6], poikiloderma, hypertrichosis (especially on the forearms), seborrhoeic dermatitis with blepharitis and eczematous dermatitis.
The ataxia of AT is cerebellar in origin and characterized by swaying of the head and trunk. Myoclonic jerks, choreoathetosis, oculomotor abnormalities and dysarthric speech become prominent in older children. Ocular apraxia, the inability to follow an object with the eyes, is a typical neurological finding. Patients are usually confined to a wheelchair by the time they are 11 years old, despite relatively preserved muscular strength. A characteristic facies develops that is hypotonic and sad in appearance. Later, the face becomes mask-like when progeric changes ensue.
Chronic and recurrent sinopulmonary infections occur in up to 80% of patients with AT and represent the most common cause of death, which is usually from resultant bronchiectasis and respiratory failure [7,8]. Other manifestations of AT include retardation of somatic growth (70%), developmental delay (30%) and endocrine abnormalities, especially ovarian agenesis or hypoplasia and insulin-resistant diabetes mellitus. Up to half of patients have evidence of glucose intolerance, hyperinsulinaemia and insulin resistance. Elevated serum transaminases are also observed in the majority of affected individuals.
Malignancy, most often a B-cell lymphoma or T-cell leukaemia, occurs in up to 40% of patients with AT who survive into their teenage years [9]. Gastric carcinomas have also been described in association with immunoglobulin A (IgA) deficiency in patients with AT, and basal cell carcinomas have been reported to develop after 20 years of age. Heterozygosity for the same ATM gene mutations that cause AT when biallelic occur in approximately 1% of the population and confer a two- to threefold increased risk of breast cancer in female carriers [10,11]. Monoallelic carriers of these mutations also have a two- to threefold increased risk of death from cancer, including malignancies of the stomach, colon and lung as well as the breast, with greater excess mortality in individuals less than 50 years of age [10].
A variety of immunological abnormalities have been described in patients with AT [7,8]. Humoral defects include decreased or absent serum levels of immunoglobulin A (IgA) (especially IgA2; in 70% of patients), IgG (especially IgG2 and IgG4; in 60% of patients) and IgE (in up to 80% of patients). Serum levels of IgM are occasionally increased, and 80% of patients have a low-molecular-weight (8S) serum IgM. Defective cell-mediated immunity occurs in approximately 70% of patients with AT and can include T- and B-cell lymphopenia, skin test anergy and deficient in vitro lymphoproliferative responses to antigens and mitogens. Patients tend to have a relative paucity of CD4+ T cells, an excess of γ/δ T cells and an absent or hypoplastic thymus. Individuals with no ATM activity have more severe immunological deficits than those with low levels of ATM activity [8].
Spontaneous chromosomal abnormalities such as fragments, breaks, gaps and translocations occur 2–18 times more frequently than normal in AT patients. Rearrangements of chromosomes 7 and 14 are especially common, and some translocations (in particular those involving 14q32) result in a proliferative advantage and may predict the development of a lymphoid malignancy. DNA in fibroblasts from patients with AT is extremely sensitive to radiomimetic agents such as bleomycin as well as to ionizing radiation.
Almost all patients with AT have elevated serum levels of carcinoembryonic antigen and α-fetoprotein, and measurement of the latter can help to establish the diagnosis in children older than 2 years of age. Radiosensitivity testing with the colony survival assay, analysis of radioresistant DNA synthesis (indicating an abnormal S-phase checkpoint), karyotyping to identify 7;14 chromosomal translocations, immunoblotting for the ATM protein, assessment of ATM kinase activity and sequencing the ATM gene may be utilized to confirm the diagnosis of AT. DNA-based prenatal diagnosis can also be performed.
Prognosis.
The median lifespan in patients with AT is 20–25 years [12]. More than half of patients die from progressive sinopulmonary disease and up to one-third succumb to malignancy or complications related to cancer treatment. Although the neurological manifestations of AT are typically progressive, some patients who survive into their thirties or forties experience stabilization of the disorder, with improvement in neurological and immunological status.
Differential Diagnosis.
The features of AT are similar to those of Nijmegen breakage syndrome (NBS), a rare autosomal recessive disorder caused by defects in the NBS1 gene (see Pathogenesis above). Nijmegen breakage syndrome features short stature, a ‘bird-like’ face, microcephaly, mental retardation and chromosomal instability. Patients with NBS have more severe immunodeficiency and a greater risk of malignancy than individuals with AT but no ataxia, telangiectasias or elevated α-fetoprotein levels. Ataxia telangiectasia-like disorder, which results from defects in the MRE11 gene (see Pathogenesis above), is characterized by radiosensitivity and neurological manifestations similar to AT but no telangiectasias, immunodeficiency, cancer predisposition or elevated α-fetoprotein levels. Lastly, the recently described RIDDLE syndrome presents with radiosensitivity, immunodeficiency, facial dysmorphism, difficulty learning, abnormal motor control and short stature resulting from defects in RNF168 protein, which mediates ubiquitin-dependent signalling at sites of DNA double-stranded breaks.
Treatment.
The treatment of patients with AT is supportive. Infections must be treated with appropriate antibiotics, and patients with severe immunodeficiency may benefit from prophylactic antibiotics and immunoglobulin replacement [13]. Avoidance of sun exposure and use of sunscreens may help to prevent actinically induced progeric changes. Physiotherapy should be instituted if there are signs of bronchiectasis, and early physical therapy may help to prevent contractures related to neurological dysfunction. Patients should be screened for the development of malignancy. Although treatment of lymphoid malignancies in AT patients with standard-dose chemotherapy is more likely to achieve remission and increase survival than reduced-dose chemotherapy, radiomimetic chemotherapeutic agents such as bleomycin and radiation therapy should be avoided. If necessary, the radiation dosage should be restricted to less than 20 Gy, in fractions of less than 1 Gy. Patients with AT have decreased thresholds for postradiation erythema, tissue necrosis and radiation-induced cutaneous malignancy.
Bone marrow and fetothymic transplants have not resulted in clinical improvement in patients with AT. Aminoglycoside-induced read-through of premature termination codons has been found to restore production of functional ATM protein in cells from individuals with truncating mutations in the ATM gene, providing the basis for a possible therapeutic approach in AT patients with this type of mutation [14]. Oxidatative stress is thought to contribute to chromosomal instability, neurodegeneration and malignancy in AT, and treatment with antioxidants such as N-acetylcysteine has been shown to decrease the frequency of lymphomas and improve motor function in Atm-deficient mice. However, in a recent pilot study of antioxidant therapy (α-lipoic acid plus nicotinamide) for patients with AT, a trend toward increased lymphocyte counts was noted but no improvement in neurological or pulmonary function was observed [13].
References
1 Savitsky K, Bar-Shira A, Gilad S et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995;268:1749–53.
2 Lavin MF. Ataxia–telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008;9:759–69.
3 Lee JH, Paull TT. ATM activation by DNA double-stranded breaks through the Mre11-Rad50-Nbs1 complex. Science 2005;308:551–4.
4 Cohen LE, Tanner DJ, Schaefer HG et al. Common and uncommon cutaneous findings in patients with ataxia telangiectasia. J Am Acad Dermatol 1984;10:431–7.
5 Paller AS, Massey RB, Curtis MA et al. Cutaneous granulomatous lesions in patients with ataxia-telangiectasia. J Pediatr 1991;119:917–22.
6 Khumalo NP, Joss DV, Huson SM, Burge S. Pigmentary anomalies in ataxia-telangiectasia: a clue to diagnosis and an example of twin spotting. Br J Dermatol 2001;144:369–71.
7 Nowak-Wegrzyn A, Crawford TO, Winkelstein JA et al. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr 2004;144:505–11.
8 Staples ER, McDermott EM, Reiman A et al. Immunodeficiency in ataxia telangiectasia is correlated strongly with the presence of two null mutations in the ataxia telangiectasia mutated gene. Clin Exp Immunol 2008;153:214–20.
9 Cabana MD, Crawford RO, Winkelstein JA et al. Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics 1998;102:98–100.
10 Thompson D, Duedal S, Kirner J et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst 2005;97:813–22.
11 Renwick A, Thompson D, Seal S et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006;38:873–5.
12 Crawford TO, Skolasky RL, Fernandez R et al. Survival probability in ataxia telangiectasia. Arch Dis Child 2006;91:610–1.
13 Lavin MF, Gueven N, Bottle S, Gatti RA. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br Med Bull 2007;81–2:129–47.
14 Lai C-H, Chun HH, Nahas SA et al. Correction of ATM gene function by aminoglycoside-induced read-through of premature termination codons. Proc Natl Acad Sci U S A 2004;101:15676–81.
Cartilage–Hair Hypoplasia Syndrome
Cartilage–hair hypoplasia (CHH) syndrome is an autosomal recessive disorder that is most common in Amish individuals and Finns. Cartilage–hair hypoplasia results from mutations in the RMRP gene encoding the RNA component of ribonucleoprotein endoribonuclease MRP, which cleaves RNA primers responsible for DNA replication in mitochondria and processes pre-rRNA in the nucleolus [1]. Patients with CHH have fine, sparse, hypopigmented hair and metaphyseal dysostosis that results in short-limbed dwarfism. Affected individuals may also have soft doughy skin with degenerated elastic tissue [2]. The majority of patients manifest some degree of defective cell-mediated immunity, which includes increased susceptibility to varicella [3,4]. Defective humoral immunity, most often deficiency of IgA or IgG, occurs in one-third of patients and may contribute to recurrent respiratory tract infections and resultant bronchiectasis [5]. Patients occasionally present with severe combined immunodeficiency (see below) and an Omenn syndrome-like phenotype of erythroderma, chronic diarrhoea, lymphadenopathy, hepatosplenomegaly and eosinophilia [4]. Anaemia (hypoplastic or autoimmune haemolytic), neutropenia (hypoplastic or autoimmune), autoimmune thyroid disease and Hirschsprung disease have also been described. Malignancy, most often non-Hodgkin lymphoma, occurs in 10% of patients. Affected individuals also have an increased risk of basal cell carcinoma [6].
References
1 Ridanpaa M, Van Eenennaam H, Pelin K et al. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, cartilage–hair hypoplasia. Cell 2001;104:195–203.
2 Brennan T, Pearson R. Abnormal elastic tissue in cartilage–hair hypoplasia. Arch Dermatol 1988;124:1411–14.
3 Notarangelo LD, Roifman CM, Giliani S. Cartilage–hair hypoplasia: molecular basis and heterogeneity of the immunological phenotype. Curr Opin Allergy Clin Immunol 2008;8:534–9.
4 Kavadas FD, Giliani S, Gu Y et al. Variability of clinical and laboratory features among patients with ribonuclease mitochondrial RNA processing endoribonuclease gene mutations. J Allergy Clin Immunol 2008;122:1178–84.
5 Toivianen-Salo S, Kajosaari M, Piilonen A, Mäkitie O. Patients with cartilage hair hypoplasia have an increased risk for bronchiectasis. J Pediatr 2008;152:422–8.
6 Taskinen M, Ranki A, Pukkala E et al. Extended follow-up of the Finnish cartilage–hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet A 2008;146A:2370–5.
Chédiak–Higashi Syndrome
Chédiak–Higashi syndrome (CHS) is an autosomal recessive disorder of vesicle trafficking characterized by mild oculocutaneous albinism, silvery hair, severe recurrent infections and haemophagocytic lymphohistiocytosis [1,2].
Pathogenesis.
Chédiak–Higashi syndrome is caused by mutations in the lysosomal trafficking regulator (LYST) gene [3]. This results in dysregulated fission/fusion of intracellular lysosome-related organelles, which leads to the formation of large dysfunctional melanosomes, platelet-dense granules and leucocyte cytolytic granules. Because the giant leucocyte granules cannot properly discharge their proteolytic and peroxidative enzymes into phagocytic vacuoles, target cell killing is ineffective. Abnormal antigen presentation, regulation of T-cell activation and repair of plasma membranes may also contribute to the pathogenesis of CHS. LYST mutations that result in a truncated protein are associated with a severe phenotype and early death, whereas missense mutations produce milder disease with survival to adulthood [4].
Clinical Features.
Patients with CHS have a characteristic silvery metallic sheen to the hair and diffuse pigmentary dilution of the skin and eyes when compared with their family members. However, affected individuals with relatively dark constitutive pigmentation often develop bronze to slate-grey hyperpigmentation acrally and in other sun-exposed sites, and superimposed guttate hypopigmented macules may also be evident in these areas [5]. Photophobia, nystagmus and strabismus can occur because of decreased ocular pigment, but visual acuity is usually normal.
Recurrent infections typically begin to develop during infancy. Infectious episodes are associated with fever and primarily involve the skin, lungs and upper respiratory tract. The most common organisms are S. aureus, Streptococcus pyogenes and Streptococcus pneumoniae. Skin infections are usually superficial pyodermas, but deeper ulcerations resembling pyoderma gangrenosum have been reported. Inflammation and ulceration of the gingiva and other oral mucosal sites may also occur. Petechiae, easy bruising and epistaxis can result from the mild bleeding diathesis of CHS.
Up to 85% of patients with CHS undergo an ‘accelerated phase’ of haemophagocytic lymphohistiocytosis that is characterized by widespread visceral infiltration with lymphoid and histiocytic cells, which are sometimes atypical in appearance. Hepatosplenomegaly, lymphadenopathy, pancytopenia, jaundice, gingivitis and pseudomembranous sloughing of the buccal mucosa are associated features. Thrombocytopenia and decreased hepatic synthesis of coagulation factors lead to increased petechiae, bruising and gingival bleeding. Infection with Epstein–Barr virus (EBV) has been implicated in the uncontrolled T-cell and macrophage activation of the accelerated phase [6].
Patients with CHS who survive into the second decade of life develop progressive neurological deterioration, which typically presents with clumsiness, an abnormal gait and paresthesias. Peripheral and cranial neuropathies, spinocerebellar degeneration, parkinsonism and dementia may complicate CHS in adults with otherwise mild disease or who underwent haematopoietic stem cell transplantation during childhood [1].
The hallmark laboratory finding of CHS is giant granules within melanocytes (melanosomes), platelets (dense granules) and leucocytes (cytolytic granules). A peripheral blood smear showing giant granules in the perinuclear area of granulocytes can help to confirm the diagnosis. Microscopically, the silvery hair of CHS displays regularly spaced clumps of melanin. Immunological abnormalities in CHS patients include neutropenia, decreased leucocyte chemotaxis, impaired natural killer (NK) cell activity, diminished antibody-dependent cell-mediated cytotoxicity, and reduced cytotoxic and regulatory T-cell function.
Prognosis.
Chédiak–Higashi syndrome is often fatal during childhood as a consequence of overwhelming infection or haemorrhage during the accelerated phase.
Differential Diagnosis.
Chédiak–Higashi syndrome should be differentiated from Griscelli syndrome (GS) (Table 177.2), a group of autosomal recessive conditions that also present with silvery hair, mild diffuse pigmentary dilution and bronzing of sun-exposed skin [7]. Griscelli syndrome is a disorder of vesicle movement and transfer that is caused by defective attachment of lysosome-related organelles to the actin cytoskeleton via a protein complex that includes myosin Va, melanophilin and Rab27A within melanocytes [8–10]. Melanocytes from individuals with GS are ‘stuffed’ with numerous melanosomes, but neutrophils have normal-appearing granules. Microscopic examination of hair shafts from patients with GS shows clumps of melanin that tend to be larger and more irregularly distributed than those seen in patients with CHS.
Table 177.2 Features of Chédiak–Higashi syndrome (CHS) and Griscelli syndromes (GS)
Adapted with permission from Bolognia JL, Jorizzo JL, Rapini RP, eds. (2007) Dermatology, 2nd edn. Elsevier, London.
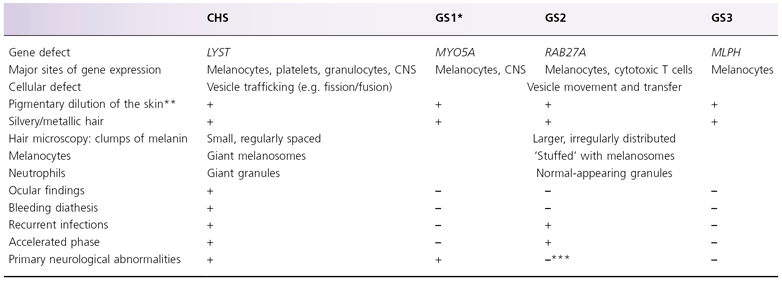
CNS, central nervous system.
GS is caused by defective attachment of organelles to the actin cytoskeleton.
* Elejalde syndrome (neuroectodermal melanolysosomal disease), which presents with pigmentary features of GS plus severe neurological dysfunction, likely represents a variant of GS1. Elejalde syndrome is not associated with immunodeficiency.
** Often accompanied by hyperpigmentation +/– guttate hypopigmented macules in acral and sun-exposed sites.
*** May develop neurological symptoms secondary to the haemophagocytic syndrome of the accelerated phase.
Type 2 GS is caused by mutations in the RAB27A gene that is expressed in T cells as well as melanocytes; like CHS, it features recurrent infections and haemophagocytic lymphohistiocytosis. Patients with type 2 GS have hypogammaglobulinaemia in addition to impaired delayed-type hypersensitivity and NK cell activity, and they may develop secondary neurological dysfunction as a complication of the accelerated phase. Type 1 GS results from mutations in the myosin Va (MYO5A) gene that is expressed in neurones as well as melanocytes, and it manifests with primary neurological abnormalities but not immunodeficiency. Elejalde syndrome (neuroectodermal melanolysosomal disease) presents with pigmentary features of GS plus severe neurological dysfunction and is thought to represent a variant of type 1 GS. Type 3 GS is caused by mutations in the melanophilin (MLPH) gene or deletion of the MYO5A F-exon, both of which are expressed primarily in melanocytes; as a consequence, the phenotype is limited to pigmentary findings.
Hermansky–Pudlak syndrome type 2 (HPS2; due to AP3B1 mutations) can present with diffuse pigmentary dilution of the skin, hair and eyes together with platelet dysfunction and recurrent infections caused by abnormal cytotoxic T and NK cells; haemophagocytic lymphohistiocytosis has also been reported [11]. However, unlike CHS, the hair is not silver in patients with HPS2. Tricho-hepato-enteric syndrome (phenotypic diarrhea of infancy) is an autosomal recessive disorder caused by mutations in the TTC37 gene that can feature diffuse pigmentary dilution of the skin and hair, immunodeficiency and platelet abnormalities; unlike CHS, it is also characterized by brittle hair with trichorrhexis nodosa, intractable diarrhea during infancy, primary liver disease, facial dysmorphism and cardiac defects [12]. Additional inherited disorders characterized by lymphoproliferation in the context of immune dysregulation are summarized in Table 177.3.
Table 177.3 Inherited disorders characterized by immune dysregulation and lymphoproliferation
Adapted with permission from Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology, 2nd edn. London: Elsevier, 2007.
AD, autosomal dominant; AR, autosomal recessive; CTL, cytotoxic T lymphocytes; EBV, Epstein–Barr virus; HSM, hepatosplenomegaly; Inh, inheritance; LAN, lymphadenopathy; LE, lupus erythematosus; NK, natural killer; XR, X-linked recessive.
Treatment.
Haematopoietic stem cell transplantation can correct the immunodeficiency of CHS and prevent or (following immunochemotherapy with etoposide, corticosteroids and ciclosporin) treat the accelerated phase [13,14]. However, transplantation does not alter the pigmentary findings or halt neurological degeneration. Management of CHS is otherwise largely supportive, with use of prophylactic antibiotics to avoid recurrent infections.
References
1 Shiflett SL, Kaplan J, Ward DM. Chédiak–Higashi syndrome: a rare disorder of lysosomes and lysosome related organelles. Pigment Cell Res 2002;15:251–7.
2 Anderson LL, Paller AS, Malpass D et al. Chediak–Higashi syndrome in a black child. Pediatr Dermatol 1992;9:31–6.
3 Barbosa MD, Nguyen QA, Tchernev VT et al. Identification of the homologous beige and Chediak–Higashi syndrome genes. Nature 1996;382:262–4.
4 Westbroek W, Adams D, Huizing M et al. Cellular defects in Chediak–Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol 2007;127:2674–7.
5 Al-Khenaizan S. Hyperpigmentation in Chediak–Higashi syndrome. J Am Acad Dermatol 2003;49:S244–6.
6 Kinugawa N. Epstein–Barr virus infection in Chediak–Higashi syndrome mimicking acute lymphocytic leukemia. Am J Pediatr Hematol Oncol 1990;12:182–6.
7 Mancini AJ, Chan LS, Paller AS. Partial albinism with immunodeficiency. Griscelli syndrome: report of a case and review of the literature. J Am Acad Dermatol 1998;38:295–300.
8 Menasche G, Pastural E, Feldmann J et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 2000;25:173–6.
9 Pastural E, Barrat FJ, Dufourcq-Lagelouse R et al. Griscelli disease maps to chromosome 15q21 and is associated with mutations in the myosin-Va gene. Nat Genet 1997;16:289–92.
10 Ménasché G, Ho CH, Sanal O et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 2003;112:450–6.
11 Enders A, Zieger B, Schwarz K et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky–Pudlak syndrome type II. Blood 2006;108:81–7.
12 Hartley JL, Zachos NC, Dawood B et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology 2010;138:2388–98.
13 Eapen M, DeLaat CA, Baker KS et al. Hematopoietic cell transplantation for Chédiak–Higashi syndrome. Bone Marrow Transplant 2007;39:411–5.
14 Trottestam H, Beutel K, Meeths M et al. Treatment of the X-linked lymphoproliferative, Griscelli and Chédiak–Higashi syndromes by HLH directed therapy. Pediatr Blood Cancer 2009;52:268–72.
Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is a group of disorders characterized by severe recurrent infections and inflammatory manifestations caused by an inability of leucocytes to kill phagocytosed organisms by generating oxidative metabolites. Reduced function of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex underlies all forms of CGD. The incidence of this disease is estimated to be 1 in 200,000 live births. The disorder usually has an X-linked recessive pattern of inheritance, but forms with autosomal recessive transmission also exist [1,2].
Pathogenesis.
The molecular defects that underlie CGD involve five components of the phagocyte NADPH oxidase:
1 Membrane-bound gp91phox encoded by the CYBB gene (70% of patients, X-linked recessive form);
2 Membrane-bound p22phox encoded by the CYBA gene (≤5% of patients, autosomal recessive form);
3 Cytoplasmic p47phox encoded by the NCF1 gene (20% of patients, autosomal recessive form);
4 p67phox encoded by the NCF2 gene (≤5% of patients, autosomal recessive form) [3]; and
5 p40phox encoded by the NCF4 gene (rare autosomal recessive form).
The membrane-bound components represent subunits of flavocytochrome b558, and upon activation of the NADPH oxidase they associate with the cytoplasmic components.
The deficient microbial killing that characterizes CGD results from failure to rapidly generate toxic reactive oxygen species (ROS) via transfer of electrons from NADPH to molecular O2 (the respiratory burst) following phagocytosis. The NADPH oxidase also has roles in activation of antimicrobial proteases within the phagosome and formation of microbicidal ‘neutrophil extracellular traps’ [4,5]. In addition to killing and degrading micro-organisms, ROS produced through NADPH oxidase activity participate in induction of neutrophil apoptosis, which normally prevents tissue damage at sites of inflammation, and regulation of cytokine synthesis. Recent studies in animal models of CGD have shown that the lack of ROS leads to excess inflammation via decreased regulatory T-cell activity, unrestrained γ/δ T-cell activity and augmented production of cytokines such as IL-8 and IL-17 [6,7].
Clinical Features.
The areas of the body that are most often affected in CGD are those that are frequently challenged by bacteria, particularly the skin, lungs and perianal region. Organisms that commonly cause infections in patients with CGD include S. aureus (especially pyoderma, abscesses and adenitis), Nocardia spp., Burkholderia cepacia, Klebsiella spp., Serratia marcescens (especially osteomyelitis), Candida spp. and Aspergillus spp. (especially pneumonia). The generation of ROS is necessary for phagocytic leucocytes to kill these organisms. The X-linked form of CGD tends to be more severe and to have a younger mean age at diagnosis (3 years) than autosomal recessive forms (8 years) [2].
The first manifestation of CGD is usually staphylococcal infections of the skin around the ears and nose in the neonatal period [8]. These localized pyodermas may progress during infancy to extensive purulent dermatitis with regional lymphadenopathy. Skin abscesses are common, especially in the perianal region. Cutaneous granulomas, which are nodular and often necrotic, occur less frequently than cutaneous infections. Purulent inflammatory reactions tend to develop at sites of minor skin trauma and heal slowly with scarring, and Sweet syndrome-like presentations have been reported [8]. Seborrhoeic dermatitis, folliculitis and ulcers of the oral mucosa (resembling aphthous stomatitis), perioral area and other cutaneous sites have also been described. Patients with CGD may develop cutaneous features of acute or chronic lupus erythematosus, especially discoid lesions [9]. In addition, female carriers of X-linked CGD occasionally present with discoid lupus erythematosus, photosensitive eruptions, Jessner lymphocytic infiltrate, Raynaud phenomenon, aphthous stomatitis and granulomatous cheilitis [10].
The extracutaneous organs typically involved in CGD are the lymph nodes, lungs, liver, spleen and gastrointestinal tract. Suppurative lymphadenitis most frequently affects the cervical lymph nodes, with abscess and fistula formation. Axillary, inguinal, mesenteric and mediastinal lymph nodes are often also involved. Pneumonia, which occurs in almost all children with CGD, responds inadequately to antibacterial therapy and can lead to abscess formation, cavitation and empyema. Hepatosplenomegaly is found in 80–90% of patients and more than one-third of affected individuals develop hepatic abscesses. Granulomas of the lungs, liver, spleen and gastrointestinal and genitourinary tracts are common and may manifest with bowel or urinary obstruction. Excessive inflammatory responses can result in additional non-infectious complications such as wound dehiscence, pneumonitis and clinical presentations mimicking inflammatory bowel disease, sarcoidosis, rheumatoid arthritis and IgA nephropathy [1,9].
Laboratory abnormalities in patients with CGD include leucocytosis, anaemia, T-cell lymphopenia, hypergammaglobulinaemia and an elevated erythrocyte sedimentation rate. Skin testing for delayed-type hypersensitivity is normal, as are studies of chemotaxis and phagocytosis. The nitroblue tetrazolium (NBT) reduction assay can be useful in establishing the diagnosis of CGD. Nitroblue tetrazolium is yellow in its soluble oxidized form; when reduced, the dye precipitates and becomes blue–black (formazan precipitate). Only 5–10% of leucocytes from patients with CGD are able to reduce NBT during phagocytosis, compared to 80–90% of leucocytes from unaffected individuals and approximately 50% of leucocytes from carriers of X-linked CGD. The dihydrorhodamine 123 assay and ferricytochrome c reduction assay are more accurate and quantitative in measuring the respiratory burst, and they can also be performed to verify the diagnosis [11].
Immunoblot analysis may demonstrate lack of the gp91phox and p22phox proteins; however, DNA analysis must be performed to ascertain which gene is affected, because mutations resulting in the absence of either protein lead to an absence of both proteins. In contrast, lack of the p47phox or p67phox protein by immunoblot analysis indicates the defective gene.
The NBT reduction and dihydrorhodamine 123 assays can be used to determine the carrier status of the sisters and other female relatives of patients with X-linked CGD, which is important to enable genetic counselling prior to pregnancy. Prenatal diagnosis of CGD using the aforementioned assays or DNA-based methods is also possible.
Differential Diagnosis.
Laboratory tests demonstrating a defective respiratory burst allow differentiation of CGD from other disorders characterized by increased susceptibility to bacterial and fungal infections. Additional inherited phagocyte disorders are summarized in Table 177.4.
Table 177.4 Inherited defects in phagocytes and Toll-like receptor signalling
Adapted with permission from Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology, 2nd edn. London: Elsevier, 2007.

See Table 25.22.2 and the text for Chédiak–Higashi syndrome and chronic granulomatous disease.
AD, autosomal dominant; AML, acute myeloid leukemia; AR, autosomal recessive; BCG, bacillus Calmette–Guérin; IFN, interferon; IL, interleukin; IRAK4, IL-1 receptor associated kinase-4; Inh, inheritance; LFA-1, lymphocyte function-associated antigen-1; MAP, mitogen-activated protein; MDS, myelodysplastic syndrome; SBDS, Shwachman–Bodian–Diamond syndrome; STAT, signal transducer and activator of transcription; TLR, Toll-like receptor; WASP, Wiskott–Aldrich syndrome protein; XR, X-linked recessive.
* Defects in STAT3 underlie AD hyper-IgE syndrome (see text), and defects in STAT5B (involved in IL-2 and growth hormone receptor signalling) lead to an AR syndrome of growth hormone insensitivity, decreased regulatory T cells, viral infections and eczematous dermatitis.
Treatment.
The use of antibiotics has markedly reduced the morbidity and mortality of CGD [12,13]. Cutaneous and nodal infections are often readily apparent. However, localized internal infections, with or without associated fever, may be difficult to detect. Thorough periodic investigation of the lungs, liver and bones by radiography, ultrasonography, computed tomography (CT), magnetic resonance imaging (MRI), positron emission tomography (PET) and bone scans often uncovers occult foci of inflammation or infection. Cultures should be performed to determine the infectious agent, and invasive procedures may be required to obtain adequate tissue samples. While awaiting culture results or in instances when culture material cannot be obtained, patients with evidence of infection should be treated empirically with broad-spectrum parenteral antibiotics that cover S. aureus and Gram-negative bacteria. Intravenous therapy should be continued for at least 10–14 days and followed by a several-week course of oral antibiotics. Surgical interventions such as débridement, irrigation and prolonged drainage may be necessary for deeper infections.
Long-term prophylactic co-trimoxazole therapy decreases the incidence of bacterial infections in patients with CGD [14]. Likewise, prophylactic administration of itraconazole diminishes the frequency of Aspergillus infections [15]. Patients with CGD have shown clinical improvement after administration of γ-interferon [16], which likely augments oxidant-independent antimicrobial pathways. Leucocyte transfusions have been used in patients with rapidly progressive life-threatening infections [13]. Short courses of systemic corticosteroids can be beneficial in the management of obstructive granulomas of the bronchopulmonary, gastrointestinal or genitourinary tracts [12,13].
Haematopoietic stem cell transplantation represents a potentially curative therapy for CGD, and its use in patients with this condition has increased in recent years [12,13,17–19]. Although younger patients without infection at the time of transplantation have better outcomes (survival >90–95%), utilization of reduced-intensity conditioning regimens has allowed successful treatment of high-risk adults with recalcitrant fungal infections. In children with CGD complicated by recurrent serious infections or corticosteroid-dependent inflammatory disease, especially those who have an HLA-identical family member, transplant should be considered before irreversible organ damage ensues.
Gene therapy was first performed in five adults with the p47phox-deficient form of CGD. A single infusion of transduced CD34+ peripheral blood stem cells led to peak levels of corrected granulocytes in 3–6 weeks, with persistence for as long as 6 months [20]. More recently, two young men with X-linked CGD were treated with non-myeloablative conditioning prior to the infusion of CD34+ peripheral blood stem cells transduced ex vivo with a retroviral vector expressing gp91phox. This led to sustained engraftment of functionally corrected phagocytes and substantial clinical improvement, with augmentation of clinical efficacy by in vivo expansion of phagocyte clones containing insertionally activated growth-promoting genes [21].
References
1 Segal BH, Leto TL, Gallin JI et al. Genetic, biochemical, and clinical features of the chronic granulomatous disease. Medicine 2000;79:170–200.
2 Winkelstein JA, Marino MC, Johnston RB et al. Chronic granulomatous disease: report on a national registry of 368 patients. Medicine 2000;79:155–69.
3 Malech HL, Hickstein DD. Genetics, biology and clinical management of myeloid cell primary immune deficiencies: chronic granulomatous disease and leukocyte adhesion deficiency. Curr Opin Hematol 2007;14:29–36.
4 Rada BK, Geiszt M, Kaldi K et al. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood 2004;104:2947–53.
5 Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol 2007;5:577–82.
6 Romani L, Fallarino F, De Luca A et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 2008;451:211–6.
7 Lekstrom-Himes JA, Kuhns DB, Alvord WG, Gallin JI. Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J Immunol 2005;174:411–7.
8 Sedel D, Huguet P, Lebbe C et al. Sweet syndrome as the presenting manifestation of chronic granulomatous disease in an infant. Pediatr Dermatol 1994;11:237–40.
9 De Ravin SS, Naumann N, Cowen EW et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol 2008;122:1097–103.
10 Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol 2007;148:79–84.
11 Jirapongsananuruk O, Malech HL, Kuhns DB et al. Diagnostic paradigm for evaluation of male patients with chronic granulomatous disease, based on the dihydrorhodamine 123 assay. J Allergy Clin Immunol 2003;111:374–9.
12 Kang EM, Malech HL. Advances in treatment for chronic granulomatous disease. Immunol Res 2009;43:77–84.
13 Seger RA. Modern management of chronic granulomatous disease. Br J Haematol 2008;140:255–66.
14 Margolis DM, Melnick DA, Alling DW et al. Trimethoprim–sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis 1990;162:723–6.
15 Gallin JI, Alling DW, Malech HL. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med 2003;348:2416–22.
16 International Chronic Granulomatous Disease Cooperative Study Group. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med 1991;324:509–16.
17 Seger RA, Gungor T, Belohradsky BH et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hematopoietic allograft: a survey of the European experience, 1985–2000. Blood 2002;100:4344–50.
18 Del Guidice I, Iori AP, Mengarelli A et al. Allogeneic stem cell transplant from HLA-identical sibling for chronic granulomatous disease and review of the literature. Ann Hematol 2003;82:189–92.
19 Gungor T, Halter J, Klink A et al. Successful low toxicity haematopoietic stem cell transplantation for high-risk adult chronic granulomatous disease patients. Transplantation 2005;79:1596–606.
20 Malech HL, Mapels PB, Whiting-Theoblad N et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci U S A 1997;94:12133–8.
21 Ott MG, Schmidt M, Schwarzwaelder K et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med 2006;12:401–9.
Chronic Mucocutaneous Candidiasis
Patients with chronic mucocutaneous candidiasis (CMC) have recurrent progressive infections of the skin, nails and mucous membranes with Candida albicans [1]. Chronic mucocutaneous candidiasis can develop in a variety of conditions characterized by ineffective immune defence against Candida. Individuals with a severely deficient immune response to Candida tend to have earlier onset and greater severity of cutaneous candidal infections. Inheritance of the autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED; also known as autoimmune polyendocrine syndrome, type I or APS1) syndrome and other familial forms of CMC is usually autosomal recessive [2]. However, many patients with CMC do not have a family history of recurrent candidal infections.
Pathogenesis.
Patients with CMC have a defective T-cell response to candidal organisms. In some individuals the deficiency is specific to Candida, and in others the immunological response to other organisms is also abnormal.
APECED results from loss-of-function mutations in the AIRE (autoimmune regulator) gene, which encodes a transcription factor [3]. The AIRE protein regulates ectopic expression of self-antigens in thymic medullary epithelial cells, thereby participating in the development of peripheral tolerance via negative selection of autoreactive T cells and generation of antigen-specific regulatory T cells [4,5]. In patients with APECED, failure to delete peripheral tissue-specific autoreactive T cells results in autoimmune disease.
Recent studies have shown that the immune defects that underlie selective predisposition to Candida infections in both APECED and non-APECED CMC patients involve impaired maturation and function of dendritic cells. In patients with APECED, stimulation of immature dendritic cells by Candida
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


