Juvenile idiopathic arthritis
Systemic lupus erythematosus
Neonatal lupus erythematosus
Juvenile dermatonyositis
Juvenile Idiopathic Arthritis
Epidemiology and Classification.
The term juvenile idiopathic arthritis (JIA) encompasses all forms of arthritis that begin before the age of 16 years, persist for more than 6 weeks and are of unknown cause [1]. The diagnosis of JIA is therefore one of exclusion and several other conditions need to be considered in the differential diagnosis (Box 175.1) [1]. JIA is the commonest paediatric rheumatic disease with an annual incidence of 10 per 100,000 children in the United Kingdom [2]. Different classification criteria have been used to identify discrete clinical subsets that could correspond to distinct diseases. The International League of Associations for Rheumatology (ILAR) has provided the most recent classification [3]. The aim of this classification was to enable identification of homogeneous groups of children with chronic arthritis to help with research on pathogenesis, epidemiology, outcome studies and therapeutic trials. The term JIA was adopted instead of juvenile chronic arthritis or juvenile rheumatoid arthritis, which were previously used in Europe and North America, respectively. Seven disease categories were recognized on the basis of features present in the first 6 months of illness (Box 175.2) [3]. This classification is based on present knowledge. However, as new information on disease pathogenesis becomes available, this classification will most likely be modified.
Box 175.1 Differential Diagnosis of Juvenile Idiopathic Arthritis (JIA) [1]
- Infection
- Other inflammatory and non-inflammatory connective tissue diseases
- Leukaemia and other malignancies, e.g. neuroblastoma
- Haemoglobinopathies
- Genetic metabolic diseases, e.g. Hurler syndrome
- Chondrodysplasias
- Autoinflammatory syndromes
Box 175.2 International League of Associations for Rheumatology (ILAR) Classification of Juvenile Idiopathic Arthritis (JIA) Subtypes [3]
Oligoarthritis
Arthritis of four or fewer joints within the first 6 months
- Persistent – affecting not more than four joints throughout the disease process
- Extended – extending to affect more than four joints after the first 6 months
Polyarthritis
Arthritis of five or more joints within the first 6 months
- RF positive – subdivided according to the presence of rheumatoid factor (RF)
- RF negative
Systemic Arthritis
Arthritis with or preceded by quotidian (daily) fever for at least 3 days, accompanied by one or more of:
- Evanescent skin rash
- Lymphadenopathy
- Hepatomegaly and/or splenomegaly conditions
- Serositis
(Mandatory exclusion of infection and malignancy; arthritis may not present early in the course)
Psoriatis Arthritis
Arthritis and psoriasis or arthritis with at least two of:
- Dactylitis
- Nail pitting or onycholysis
- Psoriasis in first-degree relative
Enthesitis-Related Arthritis
Arthritis and enthesitis or arthritis or enthesitis with two of:
- Sacroiliac joint tenderness of inflammatory lumbosacral pain
- HLA-B27 antigen
- Onset after 6 years in a male
- Acute (symptomatic anterior uveitis)
- History of HLA-B27-associated disease in a first-degree relative
Undifferentiated Arthritis
Arthritis that fulfils the criteria in no or more than two of the above categories
Pathogenesis.
The pathogenesis of JIA is still poorly understood but seems to include both genetic and environmental components. In addition, the heterogeneity of this disease indicates that different factors probably contribute to the pathogenesis. The hypothesis that an infection triggers chronic arthritis in genetically susceptible individuals is attractive, but still unproven. The results of the first genome-wide scan of children with the disease support the hypothesis that several genes, including at least one in the human leucocyte antigen (HLA) region, affect susceptibility to JIA [4]. Many associations between JIA and HLA or non-HLA molecules have been described and some have been confirmed in several studies [4,5]. Oligoarticular disease has been consistently associated with HLA antigens. Positive associations include HLA-A2, HLA-DRB1*11 (a subtype of HLA-DR5) and HLA-DRB1*08, while HLA-DRB1*04 and HLADRB1*07 have been shown to be substantially decreased [6]. Rheumatoid factor (RF)-positive polyarthritis in children has been reported to be associated, as in adults, with HLA-DR4 [7]. In enthesitis-related arthritis (ERA), 76% of patients were reported to be HLAB27 positive compared with a population frequency of around 10% [6]. A single nucleotide polymorphism (−174) in the regulatory region of the interleukin-6 gene has been associated with systemic JIA (sJIA) [8,9]. A single nucleotide polymorphism in the gene encoding tyrosine phosphatase N22 has also been associated with various autoimmune diseases, including JIA [10].
As far as the immunohistological characteristics of the synovitis in JIA are concerned, studies have revealed that the synovium shows a pronounced hyperplasia of the lining layer and a cell infiltrate that includes mononuclear cells, T cells, B cells, macrophages, dendritic cells and plasma cells [11,12]. In addition, several studies have assessed blood and synovial cytokine concentrations in children with the various subsets of JIA with inconsistent reported results [13]. The therapeutic effect of anti-tumour necrosis factor (TNF) therapies in many patients, however, supports an important pathogenic role for TNF-α.
Patients with sJIA appear to have a different inflammatory profile. Interleukin 1b (IL-1b), IL-6 and IL-18 as well as phagocytic-specific S-100 proteins (S100A8, S100A9 and S100A12) are correlated with disease activity and secondary complications [14–17]. Beside IL-6, all these molecules are secreted by a so-called alternative pathway. A loss of control of the alternative secretory pathway seems to be involved in release of proinflammatory process of sJIA [18].
Clinical Features
Systemic Arthritis
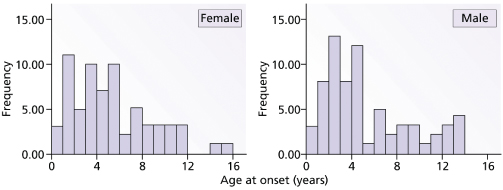
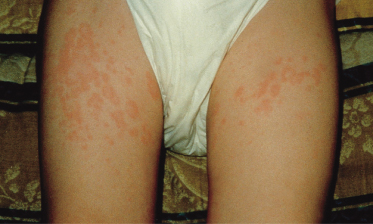
This disease typically affects young children, sometimes below the age of 1 year. Figure 175.1 shows the prevalent age of onset of systemic arthritis in the UK collected by the National Register over a 3-year period [2]. The male : female ratio is approximately the same [2]. Onset of the disease is typically one of recurrent high fevers of up to 40°C and above, often occurring at the same time of day, each day, and in between the temperature is back to normal. An evanescent macular rash, which often shows target lesions, is associated with the fever. Sometimes the rash can be urticarial and itchiness is often described by the child (Fig. 175.2). The rash usually occurs on the limbs and the cheeks, but can be all over the body and there is marked dermographism. Although the rash could be mistaken for a drug reaction or viral exanthem, the characteristic of this rash is that it is evanescent. During the fever phase, the child is acutely unwell. There is also associated hepatosplenomegaly, lymphadenopathy, serositis and often pericarditis, which can compromise cardiac function. Very often, serositis has been mistaken for an acute abdomen. Typically, arthritis occurs after the onset of fever and polyarthritis becomes prominent when the fever fades away. The time courses of these episodes are highly variable. In one follow-up study at Taplow, UK, Ansell [19] describes remission rates of 50%. The course of disease could be a single episode, or relapses with normal periods that can be months or years long, or persistent systemic features with arthritis; the latter tend to be of poorer prognosis with destructive changes of the joints. It is often said that if there is aggressive arthritis in conjunction with the onset of fever then the prognosis for these children is usually poor. This needs to be verified in prospective, multicentre studies.
Fig. 175.1 Graphs to show the prevalence of systemic juvenile idiopathic arthritis (JIA) in the UK from the National Register of the British Paediatric Rheumatology Group.
The mainstay of the management of systemic arthritis is still corticosteroid therapy, and the concurrent use of methotrexate has variable effect. The use of steroids has evolved over time. It is imperative to control the disease early and as quickly as possible to prevent cumulative side-effects and complications from the disease process. One of the possible complications is macrophage activation syndrome [20]. This is characterized by rapid clinical deterioration with features of disseminated intravascular coagulation and sometimes frank haemophagocytosis, and mortality is high. This is now the most common cause of death amongst JIA patients. In the 1950s to the 1960s, amyloidosis was the major complication and the main cause of death in this disease. Approximately 10% of sJIA patients develop this complication [21,22].
Novel therapies targeting key pathogenic molecules in sJIA have been explored recently. An initial open label, ascending dose trial of tocilizumab in severe sJIA showed it to be dramatically effective in clinical and laboratory responses observed 48 hours postinfusion [23]. Yokota et al. have reported on a double-blind, placebo-controlled, withdrawal phase III study of efficacy and safety of tocilizumab in severe sJIA, with promising results [24]. A pivotal multinational trial of tocilizumab therapy in sJIA is in progress. Furthermore, anakinra, an IL-1 receptor antagonist, has been shown to be effective in some patients with sJIA, resulting in resolution of systemic and articular symptoms [25]. Other IL-1 antagonists such as the fully human anti-IL-1β antibody canakinumab have shown good efficacy in a phase II study [1].
Oligoarthritis
This is the commonest form of JIA in children, comprising approximately 50–60% of all JIA cases. Sufferers have four or fewer joints involved at onset. Approximately 20% of these children have more joints involved in the next year or so (classified as extended oligoarthritis). The rest of the group has a better prognosis and the condition is called ‘persistent oligoarthritis’. Anterior uveitis has been found associated with this disease, in particular in younger children, typically girls around the age of 2–3 years in Europe and North America, with a positive antinuclear antibody (ANA) as an associated marker. This is not seen in other ethnic groups, such as Costa Ricans, Orientals and African black people. Typically, the course of persistent oligoarthritis is mild and remits within 2–3 years. The course of uveitis is independent of arthritis and can continue into adulthood. The extended oligoarthritis patients have a more prolonged disease course.
Polyarthritis, Rheumatoid Factor Negative
The young-onset group has similarities with oligoarthritis patients, in that they are often associated with uveitis and are positive for ANA. The drug management of this group is with methotrexate, given as a single weekly dose of 15–20 mg/m2, often as a subcutaneous injection owing to better absorption profiles. About 30% fail to respond
Polyarthritis, Rheumatoid Factor Positive
This is a small group of polyarthritis patients with symptoms that resemble adult rheumatoid arthritis in presentation, affecting mainly preteen and teenage girls. It constitutes about 1% of the Caucasian series. Young-onset polyarthritis patients are rarely rheumatoid factor positive and these patients often convert, in later years, to RF-negative polyarthritis. The drug management of these children is similar to that of RF-negative polyarthritic patients.
Enthesitis-Related Arthritis
This group of arthritis is typified by the presence of enthesitis and arthritis affecting, predominantly, the lower limb joints in boys of preteen years. The distribution of the arthritis is usually asymmetrical at onset and HLA-B27 is often positive in the Caucasian population. A proportion of patients develop sacroiliitis at puberty and these patients’ disease becomes true ankylosing spondylitis in late teenage and adulthood. Sulphasalazine has been shown to be efficacious, but not long lasting, whereas methotrexate is effective in all ages. Anti-TNF therapy is effective in those who fail to respond completely to methotrexate.
Psoriatic Arthritis
The distinguishing features of this group are the characteristic pattern of joint involvement, dactylitis, asymmetrical involvement of large and small joints and the presence of psoriasis, either in the patient or a first-degree relative. Drug treatment is similar to polyarthritis.
Treatment
The Era of Biological Therapies
The major new revolution in the management of JIA has been the advent of biological therapies, developed to block specific targets of the inflammatory response. Used successfully in patients failing conventional disease-modifying antirheumatic drugs (DMRAD), their benefit has a led to a shift in the paradigm of therapy for JIA [1]. Etanercept (soluble TNF p75 receptor fusion protein) has become an established part of managing JIA based on double-blind controlled trials of its efficacy in treating the disease. Other agents include infliximab (chimeric human/mouse monoclonal antibody that binds to soluble TNF-α), adalimumab (humanized immunoglobulin G1 monoclonal antibody that binds to TNF-α) and abatacept (soluble fully human fusion protein of cytotoxic T lymphocyte antigen 4, CTLA-4). Tocilizumab, anakinra and canakinumab are other biologic therapies for sJIA that have been extensively discussed above.
Other Treatment and Management
Physiotherapy has a major role in maintaining joint function and muscle tone during the active disease stage. This includes weight-bearing exercises as it has been shown that, if the child is not weight bearing and has hip disease, the acetabulae do not develop normally [26]. However, it is important to time the weight-bearing exercises. If the joint is acutely inflamed and under tension, it may not be a particularly good time as blood supply to the femoral head could be compromised. Intra-articular steroid therapy has been a major advance in recent years and early intervention with intra-articular steroids would be highly desirable, especially in joints such as the hip. If there are radiological changes already in the hip joint affecting the bones, intra-articular steroids could accelerate avascular necrosis. When physiotherapy and exercise fail to prevent deformity of the joints, surgical release of muscle spasm and later joint replacement therapy are all options in further management. Splinting of joints in positions of function is vital during the acute disease phase. Involvement of the chiropodist in correcting hindfoot deformity is important for normal growth of the tibia to prevent tibial torsion as well as abnormal development of the forefoot. A multidisciplinary team approach, including psychosocial input, is critical to maintain the child’s development and quality of life.
References
1 Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet 2007;369:767–78.
2 Symmons DP, Jones M, Osborne J, Sills J, Southwood TR, Woo P. Pediatric rheumatology in the United Kingdom: data from the British Pediatric Rheumatology Group National Diagnostic Register. J Rheumatol 1996;23:1975–80.
3 Petty RE, Southwood TR, Manners P et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2001;31:390–2.
4 Thompson SD, Moroldo MB, Guyer L et al. A genome-wide scan for juvenile rheumatoid arthritis in affected sibpair families provides evidence of linkage. Arthritis Care Res 2004;50:2920–30.
5 Thomson W, Donn R. Genetic epidemiology: juvenile idiopathic arthritis genetics – What’s new? What’s next? Arthritis Res 2004;4:302.
6 Thomson W, Barrett JH, Donn R et al. Juvenile idiopathic arthritis classified by the ILAR criteria: HLA associations in UK patients. Rheumatology 2002;41:1183.
7 Clemens LE, Albert E, Ansell BM. HLA studies in IgM rheumatoid-factor-positive arthritis of childhood. Ann Rheum Dis 1983;42:431–4.
8 Ogilvie EM, Fife MS, Thompson SD et al. The-174G allele of the interleukin-6 gene confers susceptibility to systemic arthritis in children: a multicenter study using simplex and multiplex juvenile idiopathic arthritis families. Arthritis Care Res 2003;48:3202–6.
9 Fishman D, Faulds G, Jeffery R et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest 1998;102:1369–76.
10 Hinks A, Barton A, John S et al. Association between the PTPN22 gene and rheumatoid arthritis and juvenile idiopathic arthritis in a UK population: further support that PTPN22 is an autoimmunity gene. Arthritis Care Res 2005;52:1694–9.
11 Murray KJ, Luyrink L, Grom AA et al. Immunohistological characteristics of T cell infiltrates in different forms of childhood onset chronic arthritis. J Rheumatol 1996;23:2116.
12 Gregorio A, Gambini C, Gerloni V et al. Lymphoid neogenesis in juvenile idiopathic arthritis correlates with ANA positivity and plasma cells infiltration. Rheumatology 2007;46:308.
13 De Benedetti F, Ravelli A, Martini A. Cytokines in juvenile rheumatoid arthritis. Curr Opin Rheumatol 1997;9:428.
14 de Jager W, Hoppenreijs EPAH, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis; a cross-sectional study. Ann Rheum Dis 2007;66:589–98.
15 Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 2005;201:1479–86.
16 Kawashima M, Yamamura M, Taniai M et al. Level of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Care Res 2001;44:550–60.
17 Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol 2007;81:28.
18 Frosch M, Roth J. New insights in systemic juvenile idiopathic arthritis from pathophysiology to treatment. Rheumatology 2008;47:121–5.
19 Ansell BM. Diagnostic criteria, nomenclature, classification. Workshop presentations and discussion. In: E Munthe, ed. The Care of Rheumatic Children. Basle: EULAR, 1978.
20 Stephan JL, Zeller J, Hubert P, Herbelin C, Dayer JM, Prieur AM. Macrophage activation syndrome and rheumatic disease in childhood: a report of four new cases. Clin Exp Rheumatol 1993;11:451–6.
21 Schnitzer TJ, Ansell BM. Amyloidosis in juvenile chronic polyarthritis. Arthritis Rheum 1997;20:245.
22 Stoeber E. Prognosis in juvenile chronic arthritis. Follow-up of 433 chronic rheumatic children. Eur J Pediatr 1981;135:225.
23 Woo P, Wilkinson N, Prieur AM et al. Open label phase II trial of single, ascending doses of MRA in Caucasian children with severe systemic juvenile idiopathic arthritis: proof of principle of the efficacy of IL-6 receptor blockade in this type of arthritis and demonstration of prolonged clinical improvement. Arthritis Res Ther 2005;7:R1281–8.
24 Yokota S, Imagawa T, Mori M et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet 2008;371:998–1006.
25 Lequerre T, Quartier P, Rosellini D et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still’s disease. Preliminary experience in France. Ann Rheum Dis 2008;67:302–8.
26 McCullough CJ. Surgical management of the hip in juvenile chronic arthritis. Rheumatology 1994;33:178.
Systemic Lupus Erythematosus
Epidemiology and Classification.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


