The relationship of autoantibody production and HLA status is also of increasing interest in scleroderma. It would appear that some of these antibodies are closely related to particular HLA alleles; for example, it has been shown that class II MHC haplotype is an important factor in determining in vitro responsiveness to topoisomerase antigen in both SSc patients and healthy control individuals [22], and linkage with HLA-DP alleles has been demonstrated [23]. These antibodies also appear to be mutually exclusive for the different subsets of scleroderma patients and, in addition, mark out within the Raynaud population those patients who are likely to develop SSc. A pathogenic role for the antibodies which target defined epitopes remains unproven. The most obvious targets for the immune response in SSc are endothelial cells and fibroblasts. It is possible that the aberrant properties of connective tissue cells (e.g. excess synthesis of collagen, fibronectin and glycosaminoglycans) and the endothelial cell damage and vasculopathy are in part consequences of immunological events in SSc. It is recognized that patients with localized scleroderma, including childhood disease, frequently demonstrate serum autoantibodies more typical of systemic connective tissue disease, including antinuclear antibodies and anti-double-stranded autoantibodies. This is intriguing as it suggests that common factors may underlie the development of autoantibodies in localized as well as systemic disease. This strengthens the view that they may represent a consequence rather than a cause of these conditions.
Whether the clinical associations of particular hallmark reactivities determined in adult SSc are also present in childhood-onset disease is uncertain. It is remarkable that anti-centromere antibody (ACA) is very rare in children whereas it is present in more than 30% of adult SSc cases. This is intriguing as many adult patients with lcSSc describe a long history of pre-existing Raynaud phenomenon that may extend back to childhood.
Cellular Interplay in Pathogenesis
The major pathological processes occurring in SSc are increased deposition of extracellular matrix (ECM) in the skin and internal organs and vascular damage. It is thought that the processes are closely related. Currently, the most favoured explanation for them implicates widespread intimal vascular damage, with endothelial cell activation, resulting in increased vascular permeability, leucocyte adhesion and subsequent emigration into the interstitium, possibly under the influence of chemotactic stimuli from resident interstitial cells. Following these events, mediators are released from the inflammatory cells and a subset of fibroblasts develops a profibrotic phenotype [24]. Whether this fibroblast abnormality is wholly acquired or whether it is dependent upon an inherited predisposition is unknown, but the final result is an excess deposition of normal matrix with subsequent organ dysfunction (Fig. 174.1).
Fig. 174.1 Interactions between endothelial cells, leucocytes and fibroblasts in scleroderma and pathogenesis. EC, endothelial cell.
Modified with permission from Oxford Textbook of Rheumatology, 2nd edn. Oxford: Oxford University Press, 1996.
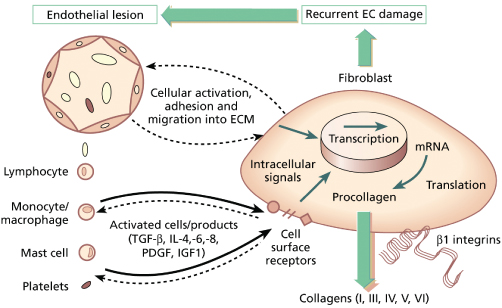
Skin fibroblasts from scleroderma patients synthesize increased quantities of many ECM proteins including collagens type I and III and to a lesser degree IV and VI, and also proteoglycan core proteins and fibronectin. Total protein synthesis and growth production are normal. The negative feedback provided by propeptides appears to be normal and these cells are not transformed or immortalized. However, scleroderma fibroblasts, unlike those from normal skin, display persistent proliferation in serum-free medium, and are without doubt deregulated with respect to ECM synthesis [25,26]. Work in the 1970s by LeRoy [27] demonstrated that fibroblasts grown from areas of sclerosis continued to synthesize increased amounts of collagen for several passages in vitro. Later, it was demonstrated that the amounts of mRNA for these matrix proteins are increased and localized predominantly to areas surrounding dermal blood vessels. Nucleic acid hybridization techniques have since confirmed that not all fibroblasts are activated, but rather that there is a subset of high collagen producers. The increased collagen RNA levels could arise through increased transcription rate, or by a reduction in mRNA breakdown, and there is evidence that both mechanisms may operate [28]. The transcriptional rate of genes encoding pro-α2(I) collagen (COL1A2) is increased in SSc fibroblasts, suggesting a change in regulatory transcription factors in these cells.
Biopsy and tissue culture studies of lesional skin in adult SSc suggest that a number of cytokines and growth factors may be involved in the initiation and/or maintenance of SSc fibroblast activation, possibly through the initiation of autocrine or paracrine loops. Thus, transforming growth factor-β (TGF-β) isoforms, connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), interleukin (IL)-1, IL-4, IL-6 and other cytokines have been identified as potential key factors [29–31].
In adult SSc, two other cell types have been implicated as key players in pathogenesis, in addition to immune cells, vascular endothelial cells and interstitial fibroblasts. Thus, the pericyte, a perivascular cell that regulates endothelial cell properties in small blood vessels, and the myofibroblast are implicated. Myofibroblasts are contractile cells derived from less specialized connective tissue fibroblasts that express a number of markers of activation and also α-smooth muscle actin. It has been suggested that myofibroblasts are responsible for much of the increased ECM production observed in adult SSc [32].
Vascular Lesions
The vascular damage in systemic sclerosis is widespread and can be recognized as Raynaud phenomenon, microvascular abnormalities with structural changes characterized by proliferative intimal arterial lesions and obliteration of the vessels leading to chronic ischaemia and intravascular pathology manifest by decreased red cell deformability, increased platelet activation and enhanced thrombus formation. The endothelial cell is thought to have the pivotal role in vascular damage in systemic sclerosis. The endothelium is now known to produce numerous molecules and to regulate many aspects of vascular stability including control of vascular tone, permeability, thrombotic potential and leucocyte trafficking [33,34].
Vasospasm in SSc is thought to arise due to changes in the levels of a variety of vasoactive mediators including nitric oxide, endothelin-1, prostacyclin, neuropeptides and platelet-released products. The mechanism underlying endothelial cell perturbation is likely to be equally complex. Both immune (cell-mediated and humoral) and non-immune cytotoxicity have been implicated [35–37]. Following injury or activation, endothelial cell expression of adhesion molecules such as E-selectin, vascular cell adhesion molecule 1 (VCAM-1) and intracellular cell adhesion molecule 1 (ICAM-1) is upregulated, usually in response to proinflammatory cytokines. These endothelial adhesion molecules bind to specific ligands on T- and B-lymphocytes, platelets, neutrophils, monocytes and natural killer cells, facilitating their adhesion to vascular endothelium and subsequent migration through leaky vessels into the ECM, where they may promote fibroblast activation. Therefore, if endothelial changes could be modulated at an early stage in SSc, it might influence the clinical expression and progression of the disease.
Clinical Features.
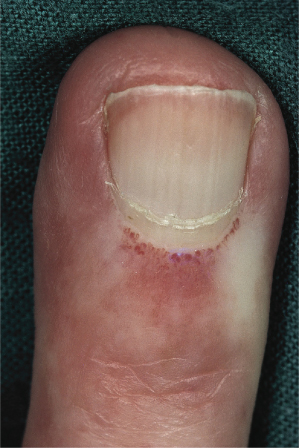
The recognition of a prescleroderma state is critical, and can be done with some accuracy. The best predictive markers of the subsequent development of scleroderma are abnormal nailfold capillaries (Fig. 174.2) and disease-specific antinuclear antibodies, and it has been suggested that the presence of both will detect over 90% of those destined to develop SSc [38,39].
Fig. 174.2 Typical macroscopic appearance of dilated abnormal nailfold capillaries in childhood-onset scleroderma.
Systemic sclerosis, once established, is a multisystem, multistage disease, and each target organ progresses through stages of inflammation, fibrosis and atrophy, not necessarily at the same time or with the same speed. The eventual effect of these processes is to impede organ function. Cutaneous involvement ( Figs 174.3, 174.4) changes in diffuse disease, usually proceeding through three phases: early, classic and late. The early stage is sometimes difficult to diagnose as the puffiness of the hands and feet can be misdiagnosed as early-onset inflammatory arthritis. A high level of suspicion is therefore needed. The face may feel slightly taut at this stage, and Raynaud phenomenon may be present. On examination, there is non-pitting oedema, with intact epidermal and dermal appendages. The subsequent, often sudden, development of classic scleroderma with firm, taut, hide-bound skin proximal to the metacarpal joints permits a definitive diagnosis in over 90% of patients. The skin may be coarse, pigmented and dry at this stage, the epidermis thins, the hair growth ceases, sweating is impaired and skin creases disappear.
Fig. 174.3 Skin sclerosis and flexion contractures in established childhood-onset systemic sclerosis. (a) Mild disease with limited skin sclerosis and mild flexion deformity. (b) More severe flexion contractures in advanced diffuse cutaneous SSc.
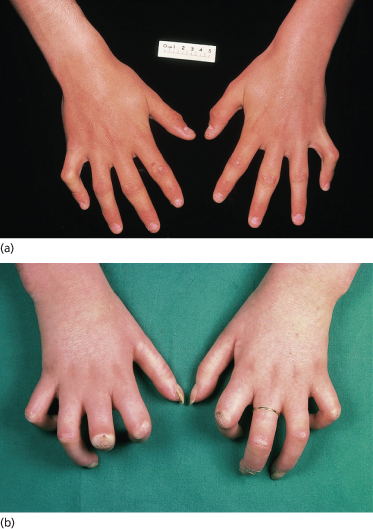
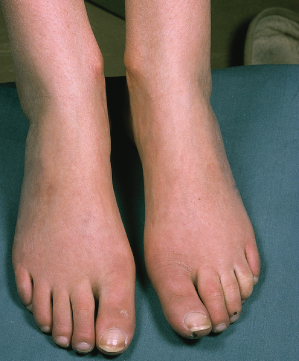
Changes limited to the fingers alone (sclerodactyly), with minimal facial and foot involvement, define the limited subset and do not carry the same implications. The classic skin changes in this group of patients, once fully developed, can remain static for many years, and the vascular aspects of the disease are usually more troublesome for the patients than the extent of skin involvement. DcSSc patients have a predominance of visceral involvement in the first 5 years of symptoms. Later, the disease often stabilizes and the skin moves into the late or atrophic phase. The taut truncal arm and leg skin may soften and, but for the pigmentation, may return almost to its normal texture. Nevertheless, the hands in patients with dcSSc nearly always show the ravages of the early active fibrotic process and contractures usually remain. Other skin manifestations include digital pitting scars, loss of finger pad pulp, ulcers, telangiectasia and calcinosis.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


