Parry–Romberg syndrome or progressive hemifacial atrophy: loss of tissue on one side of the head that predominantly involves subcutis and bone, and to a lesser extent the dermis. Overlying skin remains mobile
Disease Course and Prognosis.
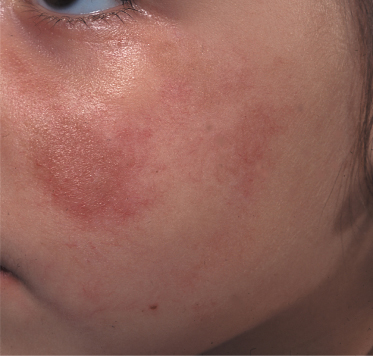
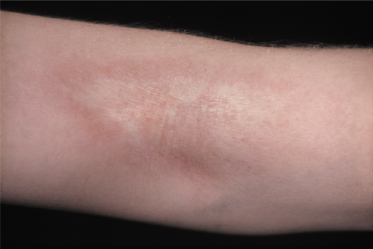
In general, morphoea starts with an early inflammatory stage, and an initial erythematous patch may be seen as the first sign of the condition (Fig. 173.1). Sometimes a telangiectatic erythema is present at this stage particularly on the face. The initial stage of the disease may be unnoticed. With spreading of the lesion and initiation of fibrosis a yellow-white elevated or depressed plaque (fibrosis) develops surrounded by a blue-violet erythema (the so-called ‘lilac ring’) (Fig. 173.2). The process develops into a more solid infiltration of the skin, resulting in atrophy with loss of hair and sebaceous glands, hyper- or hypopigmentation and an ivory-like appearance of the skin. Involvement of deeper structures varies, but usually atrophy of the underlying subcutaneous fat develops simultaneously with the cutaneous changes. The disease may additionally affect underlying muscle and bone and at the more severe end of the spectrum morphoea can result in joint contractures, extremity deformity and substantial functional and cosmetic disability [6].
The progression of morphoea varies and is dependent on the clinical type, rapidly in the case of the linear variant and more gradually with the plaque type. Plaque morphoea usually shows a mild course and is self-limiting. Generally, the clinical activity of morphoea persists for 3–4 years; new lesions may develop even after a longer time interval, but usually not after puberty [6]. However, as a child grows mildly atrophic lesions may become more evident. Softening of the lesions may occur, particularly with treatment, but complete resolution is rare. The residual inactive lesions often show hyperpigmentation. Occasionally, localized morphoea can be associated with mixed connective tissue disease with demonstrable autoantibodies; however lupus erythematosus or systemic scleroderma rarely develop [4,5].
Circumscribed or Plaque Morphoea
This variant is less common in children than in adults. It comprises two subtypes: superficial and deep. Superficial circumscribed morphoea lesions are most frequently localized on the trunk with preference for the abdominal region, especially over the iliac crests. In the initial stage the lesion shows a violaceous border, the ‘lilac ring’, and has a tendency to spread outwards. The patch has a whitish or ivory colour centrally. With time, the involved skin becomes hairless and anhidrotic, and sclerosis starts to develop [6,53]. The skin lesion becomes firmer, may show hypo- or hyperpigmentated changes and eventually signs of atrophy. Superficial circumscribed morphoea is usually associated with less physical morbity than other forms.
Dissemination of the lesions is possible and is characterized by multiple, indurated, plaque-like lesions, usually on the upper trunk, abdomen, buttocks and legs, but seldom on the face, neck and arms. Associated muscle atrophy may be observed and the joints can be involved depending on the site of the overlying skin. Some authors use the term generalized morphoea if more than four plaque-like lesions occur in two or more sites of the body [3]. However, in this author’s interpretation, generalized morphoea is characterized by widespread diffuse involvement of the body and differs from multifocal ‘en plaque’ morphoea [58].
Guttate morphoea is a term that has been used to describe multiple, small, white sclerotic lesions, which may represent multifocal circumscribed morphoea, but it needs to be differentiated from lichen sclerosus et atrophicus and atrophoderma of Pasini and Pierini. Co-existence of these diseases has been observed [59,60].
Deep, subcutaneous morphoea, referred to as ‘solitary morphoea profunda’, is localized on the upper trunk, forearms and lower legs but can also be unilateral and circular on the thigh and on the buttocks. It involves the deeper layers of the dermis and the subcutaneous fatty tissue [53,61,62], giving rise to a peau d’orange effect and sometimes ulceration. The skin is tight and immobile and arthralgia and joint contractures may result. Lesions described as morphoea profunda are likely to overlap with eosinophilic fasciitis.
Linear Morphoea of the Trunk and Limbs
Linear morphoea is the most common subtype in children, affecting approximately 65% of all paediatric patients [2]. We [49] have recently described the distribution of linear morphoea in more detail in 65 patients. Usually a limb is affected (Fig. 173.3), the legs more often than the arms. The linear lesions may sometimes affect the trunk and, rarely, half the body – face, arm, trunk and leg – resulting in hemiatrophy. In most cases linear morphoea is unilateral, and in approximately 15% bilateral lesions occur. The distribution pattern of linear morphoea has now been shown to be consistent with the lines of Blaschko, thus suggesting an early embryological development [49]. These lesions may start with an inflammatory appearance, but this is often rather subtle and may be missed. The affected skin becomes indurated and there is a change in the appearance of the skin with an indentation or puckering. Associated changes in pigmentation may vary greatly depending on the ethnic skin type of the patient. The linear variant tends to involve deeper tissues such as muscle and bone and to progress rapidly, resulting in a shortened wasted limb and joint contractures [6].
Fig. 173.3 (a) Linear morphoea affecting the right arm and hand with atrophy and contractures of the ring and small fingers. (b) Thermography of the same patient showing the affected side to be ‘hot’ or ‘active’. The increased heat detected is presumed to relate to the presence of inflammation.

‘En Coup De Sabre’ Morphoea or Linear Morphoea of the Head
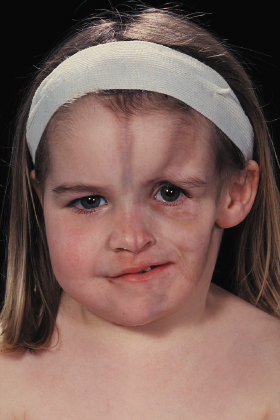
Linear morphoea of the head is termed ‘en coup de sabre’ because of the similarities of the lesions to a scar from the so-called ‘dueling sword’ injuries of the past on the forehead and scalp. ‘En coup de sabre’ morphoea in children is most often unilateral (frontoparietal or hemifacial), with a central line of demarcation [49]. After a short phase of erythema (sometimes with telangiectasia) and indurated oedema, the skin often shows early hyperpigmentation and a linear atrophic or sclerotic lesion develops, with a groove or depression [53]. The development of scarring alopecia on the scalp and partial loss of the eyebrows or eyelashes is highly characteristic. After skin and subcutis induration and atrophy, the deeper tissues may become affected with the development of facial hemiatrophy (Fig. 173.4–173.6) [63].
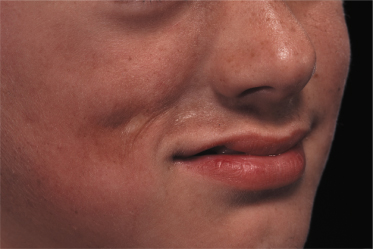
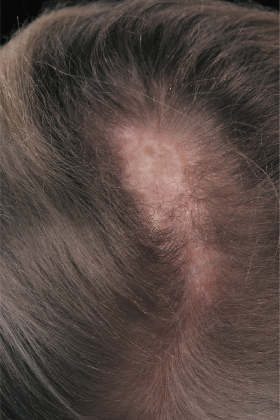
Parry–Romberg syndrome is characterized by hemifacial atrophy, mainly affecting the subcutaneous tissue, muscles and bones. In contrast to ‘en coup de sabre’ morphoea the overlying skin remains lax and movable without pigmentary changes. However, there is evidence for a close relationship and the terms Parry–Romberg syndrome and ‘en coup de sabre’ morphoea have often been used for the same clinical presentation. It is now widely accepted that Parry–Romberg syndrome represents a variant of ‘en coup de sabre’ morphoea and is not a distinct clinical entity [63,64]. These conditions share clinical, laboratorial and imaging features. The central nervous system can be affected by seizures, headache, hemiparesis and other focal neurological symptoms [63–65]. Approximately 8–13% of patients with ‘en coup de sabre’ morphoea experience seizures [2,64,65]. Magnetic resonance and/or computerized tomography imaging is useful: cerebral and skull atrophy, meningocortical alterations, intracranial calcification and white matter abnormality in the ipsilateral hemisphere can be detected [63–66]. Cerebral biopsy and analysis of cerebrospinal fluid have shown evidence of an intracerebral inflammation [67,68]. The neuroradiological findings and the response of the neurological manifestation to corticosteroids in several reports also support the hypothesis of an underlying inflammatory process [67,69].
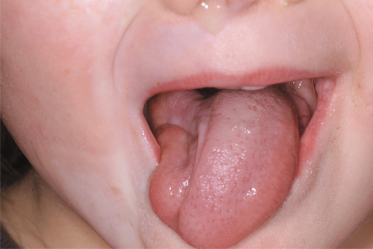
In addition, the eyes may be involved (with uveitis, globe retraction, fixed pupils and other ocular and periocular changes) [63,65] and problems affecting the jaw can occur (malalignment, disturbed dentition and difficulties opening and closing the jaw). Unilateral atrophy of the tongue can also be observed (Fig. 173.7).
Generalized Morphoea
Generalized morphoea is defined as a rare condition in which widespread and diffuse sclerosis of the skin occurs with no systemic involvement. It is mainly seen in adults. It can start insidiously, often on the trunk, with one or more plaques, and slowly progress to a much more extensive involvement. Contractures occur in limbs and can give rise to joint pains. In contrast to systemic sclerosis, Raynaud phenomenon, nail-fold capillary dilation and telangiectasia are not characteristic features. Chronic graft-versus-host disease may result in generalized morphoea [25,70].
Disabling Pansclerotic Morphoea
This rare form of morphoea initially presents as a linear variant, which rapidly progresses to affect large areas of the body [71,72]. First described in 1923 [73] as ‘progressive mutilating scleroderma’, the condition is characterized by a polymorphous appearance of lesions with involvement of the skin, deep structures, tendons, fascia and muscles. Arthralgia, joint stiffness, contractures of the hands and extremities and skin ulcerations are common features. The histological abnormalities include panniculitis and a marked lymphocytic inflammation [74]. Systemic involvement in the form of lung fibrosis and oesophageal dysmotility have been described and the condition is fatal in some cases [72,74]. Recent reports have raised awareness of the possible evolution of chronic skin ulceration to squamous cell carcinoma in pansclerotic morphoea of childhood [75,76].
Mixed Morphoea (Combined Forms)
Combined forms of morphoea are seen in approximately 14–15% of paediatric patients [2,8]. Most often combinations of circumscribed lesions on the trunk with a linear lesion on the leg or an ‘en coup de sabre’ lesion are seen. The overlap is indicative of a common pathophysiology and demonstrates the difficulty in classification on solely clinical grounds. The combined types tend to be more aggressive and usually require systemic treatment. Other combined forms are: morphoea, lichen sclerosus et atrophicus and atrophoderma of Pasini and Pierini.
Extracutaneous Involvement and Associated Conditions
Morphoea is usually limited to the skin and subcutaneous tissue and, in general, internal organs are not involved. However, extracutaneous involvement is present in approximately 22% of patients, as recently described in a multicenter study of 750 children with morphoea [4]. Extracutaneous manifestations were more frequently seen in patients with linear morphoea. The extracutaneous manifestations were as follows: musculoskeletal (particularly articular) involvement 19%; neurological symptoms (including seizures, deafness, transient ischaemic attacks, migraine and headaches) 4%; other autoimmune conditions (including Raynaud phenomenon) 3%; vascular findings (vasculitic rash and deep vein thrombosis) 2%; ocular involvement 2%; and gastrointestinal symptoms (e.g. gastro-oesophageal reflux) and respiratory findings (restrictive lung disease) 1%. Very rarely cardiac findings (pericarditis and arrhythmia) have been described. The bone changes in the form of thinning, hypoplasia and shortening, as seen in linear morphoea, are usually secondary, however arthritis and musculoskeletal symptoms may occur unrelated to the site of the skin lesion.
Extracutaneous involvement seems to be associated with a positive rheumatoid factor, circulating ANAs, elevated erythrocyte sedimentation rate and C-reactive protein [4]. In these patients the extracutaneous involvement is milder and not life threatening as usually seen in patients with systemic sclerosis. If more severe complications occur, such as pulmonary disease, pulmonary hypertension, cardiomyopathy and generalized myopathy, they should be interpreted as a sign of systemic sclerosis [77,78].
Other autoimmune syndromes may be associated with morphoea; however this is less frequent in children (3–5%) than in adults (30%) [4,23]. The most common associated autoimmune disorders include vitiligo, alopecia areata, lichen planus, psoriasis, inflammatory bowel disease, type 1 diabetes mellitus and connective tissue diseases such as discoid and systemic lupus erythematosus, dermatomyositis. Sjögren syndrome and rheumatoid arthritis [23].
Laboratory Abnormalities.
There may be an increase in C-reactive protein in childhood-onset morphoea (in approximately 9%), but this is usually not paralleled by other acute phase response markers, including erythrocyte sedimentation rate, immunoglobulin and complement [2,12]. Eosinophilia may occur in 7–18% [2,5,8] and reduced complement C2 has been reported [79].
ANA, anti-Ro/SSA and rheumatoid factor are often found in children with morphoea, the clinical relevance of which is uncertain. In recent larger cohorts of patients, ANAs were found in 26–59% [2,5,8]. This frequency in children is lower than in adults with localized scleroderma but higher than in the normal population. Linear, deep and generalized morphoea seem to be the subtypes associated with a higher prevalence of ANA; however, no correlation between these antibodies and the disease course has been observed [2,8]. One of the major autoantigens for ANAs in morphoea is nuclear histone. Antihistone antibodies (AHAs) have been detected in 47% of patients with morphoea, with a different prevalence in the various subtypes – higher in generalized morphoea, lower in circumscribed morphoea [80]. Monitoring AHA titres has been proposed for assessing disease activity, however this has not been further studied in larger cohorts [81].
Anti-topoisomerase I antibodies (anti-Scl 70), a marker for systemic sclerosis, are rarely detected in children with morphoea (2–3%) [2,23,80]. Anticentromere antibodies (ACAs) are found more commonly in adult patients (12%) than in children with morphoea (1.7%) [2,82]. Whether these antibodies are markers that reflect the immunological component of the disease process or can have a prognostic significance is unclear. None of the Scl-70- or ACA-positive patients in the series of 750 morphoea cases developed signs or symptoms of internal organ involvement during a mean follow-up of 3.4 years [2]. Rheumatoid factor, a serological marker for rheumatoid arthritis, has been detected in approximately 15% of paediatric patients with morphoea and seems to correlate with the presence of arthritis [2]. A recent study underlined the role of anti-DNA topoisomerase IIα (antitopo-IIα) autoantibodies in morphoea. These antibodies were detected in 76% of patients with morphoea, whereas in only 14% of patients with systemic sclerosis [83]. Although it remains unknown why morphoea is associated with antitopo-IIα but not antitopo-I antibodies and vice versa in systemic sclerosis, these antibodies seem to play an important role in fibrotic disorders as shown by their presence in idiopathic pulmonary fibrosis [84]. Antibodies against anticardiolipin and lupus anticoagulant have also been found in paediatric morphoea patients with a prevalence of approximately 12%, which is lower than that reported for adult patients (46%) [2,85].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


