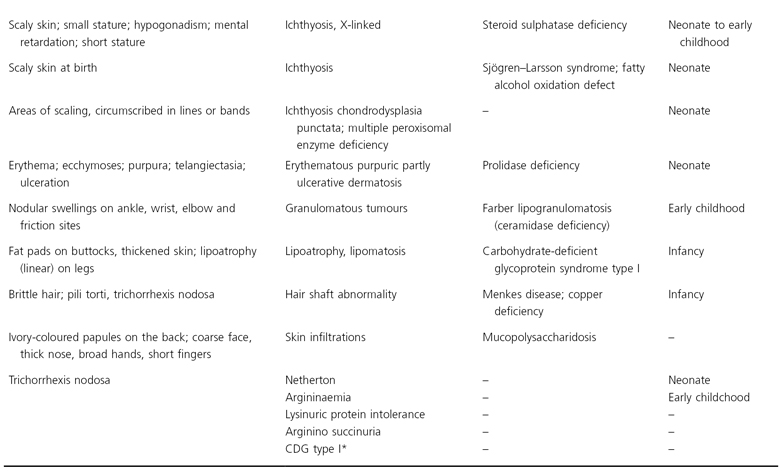
* Silengo M, Valanzise M, Pagliardini S et al. Hair changes in congenital disorders of glycosylation (CDG type I). Eur J Pediatr 2003; 162: 114–15.
References
1 Saudubray JM, Charpentier C. Clinical approach to inherited metabolic diseases. In: Scriver CR, Beaudet AL, Sly WS et al. (eds) The Metabolic and Molecular Bases of Inherited Disease. Clinical Phenotypes: Diagnosis/Algorithms, 8th edn. New York: McGraw-Hill, 200, pp. 1327–407.
2 Saudubray JM, Ogier de Baulny H, Charpentier C. Clinical approach to inherited metabolic diseases. In: Fernandes J, Saudubray JM, van den Berghe G (eds) Inborn Metabolic Diseases: Diagnosis and Treatment, 3rd revised edn. Berlin: Springer Verlag, 2000, pp. 3–41.
3 Saudubray JM, Ogier H, Bonnefont JP et al. Clinical approach to inherited metabolic disease in the neonatal period: a 20-year survey. J Inher Metab Dis 1989;12(Suppl 1):25–41.
Aminoacidopathies
Phenylketonuria/Hyperphenylalaninaemia
Definition.
Phenylketonuria (PKU) is an autosomal recessively inherited aminoacidopathy, caused by deficiency of L-phenylalanine hydroxylase in the liver [1].
History.
In 1934, Fölling [2] described 10 patients with oligophrenia with a typical dermatitis on air-exposed skin, muscle hypotonia and green coloration of the urine after addition of ferrichloride. He named this condition imbecillitas phenylpyruvica because of the abnormal amount of phenylpyruvic acid in the urine.
In 1937, this condition was named oligophrenia phenylpyruvica by Jervis [3], indicating the different clinical stages of this disease. In 1953, treatment with a phenylalanine-restricted diet by using casein hydrolysate was described by Bickel et al. [4].
Pathogenesis.
Phenylketonuria is a genetically determined disease with an autosomal recessive inheritance. A small group of patients (1–2%) with hyperphenylalaninaemia (HPA) have defects of biopterin synthesis metabolism that may result in a deficiency of neurotransmitters. Owing to the neurological sequelae, the name ‘malignant PKU’ is used for this group [5].
The genetic incidence of PKU varies between countries; over 400 mutations in the phenylalanine hydroxylase gene have already been described. In Ireland, its frequency is high (1 in 400 births); in Finland very low (1 in 40,000 births).
Clinical Features.
In the more severe classic untreated or late detected forms of PKU (plasma phenylalanine concentrations ≥1200 µmol/L (normal values 60 µmol/L)), retarded development, seizures, microcephaly and mental retardation are constant clinical features.
The condition is characterized by decreased skin pigmentation, photosensitivity, eczema, lightly pigmented eyes, fair hair and sclerodermoid changes. Synthesis of brain neurotransmitters is reduced owing to competitive inhibition of tyrosine and tryptophan hydroxylation. Patients with mild PKU/HPA (plasma phenylalanine levels ≥240 µmol/L but not over 480 µmol/L) reflect a less severe clinical phenotype.
Untreated pregnant women with HPA/PKU have a high risk of spontaneous miscarriage (maternal PKU syndrome); their offspring often show low birthweight, microcephaly, retarded development and cardiac malformations [6,7].
Prognosis.
In classic PKU, early dietary treatment with a phenylalanine-restricted diet can prevent psychomotor delay and mental retardation.
Patients with defective biopterin biosynthesis should be carefully monitored for neurological symptoms. Some patients with these defects experience a rather benign course; however, some show severe neurological sequelae even if well treated. The long-term prognosis of these disorders is still unknown. Prevention of maternal PKU syndrome will be a main issue in the near future, as the generation of patients whose condition was detected early by newborn screening has now reached adulthood.
Differential Diagnosis.
In the neonatal period and in premature infants and patients with liver disease, plasma levels of phenylalanine can be abnormally high. This can lead to a false-positive PKU screening test due to other metabolic defects, such as classic galactosaemia or tyrosinaemia (see below). The dermatological manifestations in PKU can mimic eczema and scleroderma-like lesions with pigment changes.
Screening and Treatment.
In the USA and most western countries, PKU screening is included in the national neonatal screening programme using either a semi-quantitative microbiological procedure (Guthrie card) or a quantitative chemical reaction (Quantase test) [8].
A low-phenylalanine, tyrosine-supplemented diet should be introduced as soon as possible if plasma phenylalanine concentrations exceed 360 µmol/L. Vitamins, minerals and calcium should be added according to need and age. All patients should remain on this diet for life, or at least until they leave school; women of child-bearing age should remain on the diet to prevent maternal PKU syndrome. Some centres institute a protein-restricted diet in adulthood. During treatment, patients’ hair colour may darken as a result of tyrosine supplementation in the diet (Fig. 169.1).
Fig. 169.1 Change in hair pigmentation after the start of a phenylalanine-restricted, tyrosine-supplemented diet in a late-detected classic phenylketonuria patient.
Courtesy of Prof. F.J. van Sprang.

In patients with defects in biopterin metabolism, administration of tetrahydrobiopterin (BH4) and precursors of neurotransmitters (L-dopa, carbidopa and 5-hydroxytryptophan) is the treatment of choice, depending on the enzymatic defect. In the last decade BH4– dependent classic PKU has been shown as a new entity; 20 mg/kg daily of BH4 reduces the plasma phenylalanine by 30%, allowing a reduction of the protein-restricted diet and a better quality of life [9,10].
Tyrosinaemia
Definition
Tyrosinaemia type I is the hepatorenal type caused by fumarylacetoacetase deficiency [11]. No skin abnormalities are present. Tyrosinaemia type II (Richner–Hanhart syndrome) is the oculocutaneous form of tyrosine catabolism due to a defect of hepatic cytosol aminotransferase [12]. Tyrosinaemia type II is an autosomal recessive disorder.
History.
Richner in 1938 and Hanhart in 1947 recognized a distinctive oculocutaneous syndrome. Its association with tyrosinaemia was suggested 35 years after the original clinical description. Many patients have probands of Italian ancestry, although patients have been described from all over the world.
Pathogenesis.
Oculocutaneous tyrosinaemia or Richner–Hanhart syndrome is one of the known inborn errors of tyrosine catabolism, and is due to a defect of hepatic cytosol aminotransferase. This results in increased plasma tyrosine levels as well as in abnormal urinary tyrosine metabolites.
Alkaptonuria, another tyrosine-related disorder, characterized by dark-stained urine and cerumen, and arthritis and ochronosis in the fourth decade of life, is discussed below.
Intracellular crystallization is proposed as the mechanism for tissue damage. The levels of amino acids in the epidermis may exceed those in plasma, as tyrosine levels in plasma exceed saturation. The skin lesions are limited to volar surfaces and are associated with polymorphonuclear infiltrates.
Clinical Features.
Tyrosinaemia type I presents in the neonatal period as an acute disease with severe liver failure or as a more chronic form later in life with rickets, failure to thrive, porphyria-like attacks and liver failure [11]. No skin abnormalities are present. Treatment with a chemical drug, NTBC (Nitisinone, Orfadin), prevents hepatocellular carcinoma and the need for liver transplantation and provides a better life expectancy, although a special diet is still necessary.
Oculocutaneous tyrosinaemia or Richner–Hanhart syndrome is characterized by skin abnormalities showing painful palmar and sole erosions and hyperkeratosis. These symptoms often begin as painful blisters. The eye lesions are often observed within the first year (but usually within the first decade) as bilateral erosions with photophobia or therapy-resistant keratitis. As a result, visual impairment can occur if correct diagnosis and treatment are delayed. Tyrosine crystals in the eye can also be observed on slit-lamp examination. The basis for the neurological symptoms often seen in this disease is not known.
Prognosis.
Dietary restriction of phenylalanine and tyrosine can resolve the ophthalmological abnormalities within a week. The skin lesions will recover within a few months. Pregnancy in patients with tyrosinaemia type II has been described [13].
Differential Diagnosis.
Painful blisters in oculocutaneous tyrosinaemia should be considered as a differential diagnosis of various blistering diseases, such as different forms of epidermolysis bullosa.
Treatment.
Oculocutaneous tyrosinaemia with eye crystals is treated with a phenylalanine- and tyrosine-restricted diet (the diet should also be low in phenylalanine because the conversion from the amino acid phenylalanine to tyrosine by L-phenylalanine hydroxylase is not hampered). Dietary intervention is often successful. On dietary treatment, neurological symptoms generally disappear [14].
Homocystinuria
Definition.
Cystathionine β-synthase (CBS) deficiency is a genetic disorder of trans-sulphuration [15].
History.
The enzyme defect was identified in 1964 by Mudd et al. after the first clinical description in 1963 by both Carson and Neill in Northern Ireland and Gerritsen et al. in the USA [15].
Pathogenesis.
Cystathionine β-synthase deficiency is a disorder of trans-sulphuration resulting in elevated plasma levels of homocyst(e)ine and methionine and a decreased level of cysteine [15]. CBS deficiency is inherited as an autosomal recessive trait; pyridoxine responsiveness is probably related to mutations of specific regions of the protein.
Clinical Features.
Affected patients have a multisystem involvement, including eyes (ectopis lentis), skeleton (marfanoid habitus, premature osteoporosis, bone deformities, e.g. pectus carinatum, pectus excavatum), central nervous system (mental retardation), vascular system (thromboembolic events), skin (malar flush) and hair.
Reversible hypopigmentation in some treated homocystinuric patients has been reported; the mechanism is undefined, although some data suggest tyrosinase inhibition by homocyst(e)ine caused by interaction of homocyst(e)ine with copper at the active site of tyrosinase [16]. Malar flush in untreated patients manifests as roughening and red coloration of the skin of the cheeks, as seen in farmers (Fig. 169.2).
Fig. 169.2 Three Moroccan siblings with CBS (cystathionine β-synthase-deficient pyridoxine-responsive homocystinuria) before treatment. The boy’s lenses have been extracted; the elder girl shows the facial malar flush.

Prognosis.
Clinical variability is present and mild cases may not be recognized until severe complications, such as thromboembolic accidents, develop. Even mild hyperhomocyst(e)inaemia has been related to higher risk of occlusive vascular disease, partly related to methylenetetrahydrofolate metabolism.
Differential Diagnosis.
The classic phenotype is often confused with Marfan syndrome, an autosomal dominantly inherited disease, and with eye lens dislocation, skeletal involvement and vascular complications (aneurysma of the aorta). Homocystinuria and Marfan syndrome are both systemic diseases with (different) collagen abnormalities. Mental retardation is not associated with Marfan syndrome, unlike homocystinuria.
Treatment.
About half of the patients with homocystinuria due to CBS deficiency respond to pharmacological doses of pyridoxine (three doses at 100–250 mg daily orally). Pyridoxal 5-phosphate is a co-factor for CBS, and pyridoxine treatment (vitamin B6) will increase the conversion of pyridoxine non-responsive homocysteine to cysteine in patients with CBS deficiency. Betaine (dimethylglycine, as a methyl donor) is also used in the treatment of homocystinuria to stimulate the remethylation of homocysteine to methionine [17,18].
Other causes of (mild) hyperhomocysteinaemia can be caused by folic acid or vitamin B12 deficiency, or by inborn errors of cobalamin like Cbl C, D or F which are combined with methylmalonic acidurias.
References
1 Güttler F. Hyperphenylalaninaemia. Diagnosis and classification of the various types of phenylalanine hydroxylase deficiency in childhood. Acta Pediatr Scand 1980;280(Suppl):1–80.
2 Fölling A. Uber Ausscheidung von Phenylbrenztraubensaüre in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität. Höppe-Seylers Z Physiol Chem 1934;227:169.
3 Jervis GA. Phenylpyruvic oligophrenia. Arch Neurol Psychiatr 1937;38:944.
4 Bickel H, Gerrad I, Hickmans EM. Influence of phenylalanine intake on phenylketonuria. Lancet 1953;ii:812.
5 Blau N, Thony B, Heizmann CW et al. Tetrahydrobiopterin deficiency: from phenotype to genotype. Pteridines 1993;4:1–10.
6 Lenke RL, Levy HL. Maternal phenylketonuria and hyperphenylalaninaemia. N Engl J Med 1980;303:1202–8.
7 De Klerk JBC, Wadman SK, Dijkhuis HJ et al. Maternal PKU syndrome in an exceptional family with unexpected PKU. J Inher Metab Dis 1987;10:162–70.
8 Smith I, Cook B, Beasley M. Review of neonatal screening programme for phenylketonuria. BMJ 1991;303:333–5.
9 Blau N, Erlandsen H. The metabolic and molecular bases of tetrabiopterin-responsive phenylalanine hydroxylase deficiency. Mol Genet Metab 2004;82:101–11.
10 Muntau AC, Roschinger W, Habich M et al. Tetrabiopterin as an alternative treatment for mild phenylketonria. N Engl J Med 2002;347:2122–32.
11 Kvittingen EA. Tyrosinaemia type I – an update. J Inher Metab Dis 1991;14:554–62.
12 Paige DG, Clayton P, Bowron A et al. Richner–Hanhart syndrome (oculocutaneous tyrosinaemia type II). J Roy Soc Med 1992;85:759–60.
13 Francis DEM, Kirby DM, Thompson GN. Maternal tyrosinaemia type II. management and successful outcome. Eur J Pediatr 1992;151:196–9.
14 Barr DGD, Kirk JM, Laing SC. Outcome of tyrosinaemia type II. Arch Dis Child 1991;66:1249–50.
15 Mudd SH, Skovby F, Levy HL et al. The natural history of homocystinuria due to cystathionine β-synthase deficiency. Am J Hum Genet 1985;94:1–31.
16 Rish O, Townsend D, Berry SA et al. Tyrosine inhibition due to interaction of homocysteine with copper: the mechanism for reversible hypopigmentation in homocystinuria due to cystathionine β-synthase deficiency. Am J Hum Genet 1995;57:127–32.
17 Mudd SH, Levy HL, Krans JP. Disorders of trans-sulfuration. In: Scriver CR, Beaudet AL, Sly WS et al. (eds) The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill, 2001, pp. 2007–56.
18 Wilcken DEN, Wilcken B, Dudman NPD et al. Homocystinuria – the effect of betaine in the treatment of patients not responsive to pyridoxine. N Engl J Med 1983;309:448–53.
Organic Acidurias
Alkaptonuria
Definition.
Alkaptonuria is an autosomal recessive hereditary disease in which the enzyme homogentisic acid oxidase is absent. Owing to this effect, homogentisic acid accumulates and is excreted in urine.
History.
Studies on alkaptonuria have been of great importance in the development of ideas about inherited metabolic diseases. In 1908, Sir Archibald Garrod discussed alkaptonuria in one of the Croonian lectures [1].
Pathogenesis.
As a result of homogentisic acid oxidase enzyme deficiency, homogentisic acid accumulates and is excreted in the urine. Homogentisic acid is an intermediary product in the metabolism of the aromatic amino acids phenylalanine and tyrosine.
Clinical Features.
If urine contains homogentisic acid, the acid is gradually oxidized to a melanin-like product, resulting in dark-stained urine. This may lead to early recognition of the disease (black–brown darkening of used napkins). The main features of alkaptonuria are signs due to the presence of homogentisic acid in urine, darkened cerumen of the ears, pigmentation of cartilage, generalized pigmentation in connective tissues (ochronosis) and, later in life, arthritis and bluish pigmentation of the sclera. The skin manifestations are the first diagnostic signs.
Prognosis.
Arthropathy occurs in the major joints and spine in the third and fourth decades. Calcification in the heart valves has been described. In comparison with the general population, a higher incidence of cardiovascular disease due to arteriosclerosis is described.
Differential Diagnosis.
A familial and specific intestinal malabsorption of tryptophan, known as ‘blue napkin syndrome’, has been described [2]. Unabsorbed tryptophan is converted to indican by intestinal bacteria (which can also mimic Hartnup disease). The bluish pigmentation is suggestive of haemochromatosis or naevus of Ito. The same pigmentation can be observed after local treatment with phenolic solutions. Chronic arthritis as a manifestation of long-standing alkaptonuria resembles gouty arthritis or rheumatoid arthritis.
Treatment.
Several attempts to treat alkaptonuria have been studied (vitamin C, vitamin B12, cortisone) without any clinical effect. Dietary restriction of phenylalanine and tyrosine (or general protein restriction) seems to be logical in order to reduce the production of homogentisic acid. The final outcome, however, is unclear. The symptomatic prevention of complications (joints, heart) seems to be the primary therapeutic goal.
Administration of NTBC [2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione], as in tyrosinaemia type I, could reduce homogentisate formation and thus prevent complications. The cost of and experience with this orphan drug, however, make its long-term use uncertain [3].
Propionic Acidaemia
Definition.
Propionic acidaemia, along with methylmalonic acidaemia, is an autosomal recessively inherited inborn error of branched-chain amino acid metabolism.
History.
In 1961, Childs described an infant with episodic metabolic ketoacidosis, protein intolerance and elevated plasma glycine concentrations. Many children with similar clinical and biochemical findings have since been described. Propionic acidaemia due to a deficiency of propionyl co-enzyme A (CoA) carboxylase deficiency and different forms of methylmalonic acidaemia, all with markedly elevated plasma glycine concentrations, fit this description.
Pathogenesis.
The essential amino acids, isoleucine, valine, methionine and threonine, are metabolized into propionyl CoA. Propionyl CoA is converted into methylmalonyl CoA by the enzyme propionyl CoA carboxylase. Deficiency of this enzyme causes propionic acidaemia [4].
Clinical Features.
The clinical characteristics of propionic acidaemia and methylmalonic acidaemia are similar. Patients are commonly seen in the first weeks of life with vomiting, metabolic acidosis, failure to thrive, hypotonia and mental retardation: neonatal death occurs.
Propionic acidaemia is one of the so-called ketotic hyperglycinaemia syndromes and is characterized by increased concentrations of free propionate in blood and urine. 1-Methylcitrate and 3-hydroxypropionate are major diagnostic urinary metabolites. During decompensation, plasma levels of odd-numbered fatty acids, ammonia and glycine are elevated.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


