Wegener granulomatosis
Polyarteritis nodosa and microscopic polyangiitis
Behçet disease
Relapsing polychondritis
Wegener Granulomatosis
Wegener granulomatosis (WG) is one of the antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV) which also include microscopic polyangiitis (MPA, formerly called microscopic polyarteritis), Churg–Strauss syndrome (CSS) and so-called renal-limited vasculitis (previously referred to as idiopathic crescentic glomerulonephritis) [1].
Wegener granulomatosis is defined according to the Chapel Hill criteria as granulomatous inflammation involving the respiratory tract, with necrotizing vasculitis affecting small to medium-sized vessels (e.g. capillaries, venules, arterioles and arteries); necrotizing glomerulonephritis is common [2].
Wegener granulomatosis was first described by Friedrich Wegener in 1936, based on three similar autopsy cases. Nearly 20 years later, Godman & Churg named it the ‘Wegener triad’ [3]. It is a multisystem disease affecting the ears, nose, larynx, eyes, lungs, kidneys, heart, joints, skin and central nervous system (CNS) [4]. It is predominantly a disease of adults, usually 40–50 year olds, with a male-to-female ratio of 1.3–1.6 : 1. However, the disease does occur in children, and of the AAV occurring in the young, WG is seen most frequently [5].
The recently modified classification for WG requires the presence of three out of the following six criteria:
- renal involvement (proteinuria or haematuria or red blood cell casts)
- positive histopathology (granulomatous inflammation within the wall of an artery or in the perivascular or extravascular area)
- upper airway involvement (nasal discharge or septum perforation, sinus inflammation)
- laryngo-tracheo-bronchial involvement (subglottic, tracheal or bronchial stenosis)
- pulmonary involvement (chest X-ray or CT scan)
- ANCA positivity (by immunofluorescence, ELISA PR3-ANCA or MPO-ANCA).
Sensitivity and specificity of these new EULAR/PRINTO/PRES criteria are 93% and 99% respectively for the differentiation of WG from other forms of vasculitis in the young [5].
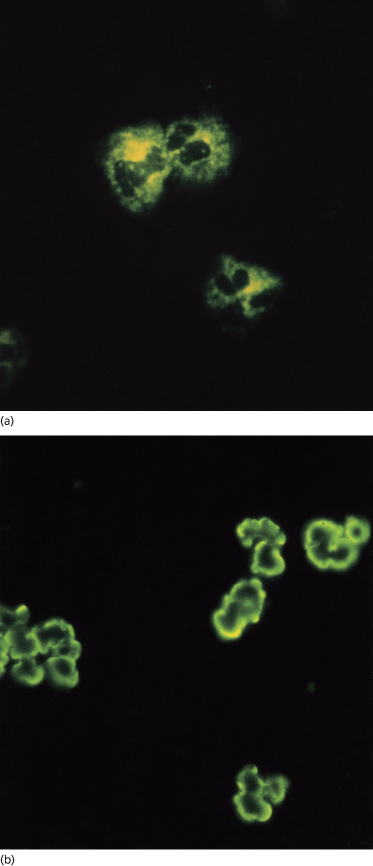
The diagnosis of AAV can be challenging and ANCA undoubtedly play an important role. Davies et al. in 1982 [6] described antibodies directed against antigenic determinants in the neutrophil cytoplasm of patients with segmental necrotizing glomerulonephritis. Methodological improvements for the detection of ANCA have resulted in increased sensitivity and specificity. Both indirect immunofluorescence (IIF) and enzyme-linked immunosorbent assay (ELISA) are used for routine diagnostic purposes. Typically, WG is associated with a cytoplasmic staining pattern of ANCA on IIF, and ELISA reveals specificity against proteinase 3 (PR3-ANCA). However, perinuclear ANCA (pANCA) with specificity directed against myeloperoxidase (MPO-ANCA) can also be found in patients with WG (Fig. 167.1). MPA and renal-limited AAV are typically associated with pANCA on IIF and MPO-ANCA on ELISA. Lastly, it should always be remembered that ANCA-negative forms of WG, MPA, renal-limited vasculitis and CSS are well described in children [5].
Other commonly observed non-specific findings include a mild normochromic normocytic anaemia together with a leucocytosis and thrombocytosis. The erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are frequently elevated. Raised immunoglobulins (polyclonal IgG) may also support the diagnosis, although this finding is not specific. Laboratory manifestations relating to renal involvement include dipstick haematuria and proteinuria, a raised protein creatinine ratio, raised serum creatinine and other associated laboratory features of renal failure.
The presence of ANCA (especially cANCA) has been found by some authors to be highly specific for WG, with up to 91% sensitivity and 98% specificity during active disease being reported in adults [7,8]. In children, there is also evidence to show a significant association with ANCA. In one report 10/12 patients had ANCA detected although only seven had the cANCA pattern on immunofluorescence [9]. In subsequent reports, ANCA positivity was found in 59–95% [10,11]. It is likely that this range of ANCA positivity in children with WG reflects differences in age, variation in disease severity, and methodological differences in laboratory detection of ANCA between these two series. Whilst early reports by Van der Woude et al. [12] demonstrated the presence of these antibodies in adults with WG and correlated their presence with disease activity, more recent studies have questioned the usefulness of measuring ANCA to monitor disease activity [13] (see below).
Aetiology and Pathogenesis.
The most accepted model of pathogenesis proposes that ANCA activate cytokine-primed neutrophils, leading to bystander damage of endothelial cells and an escalation of inflammation with recruitment of mononuclear cells [14]. However, other concomitant exogenous factors and genetic susceptibility appear to be necessary for disease expression as well.
The aetiology of ANCA generation in WG is unknown. It is clear that infections may precipitate the onset of the disease or relapses, perhaps inducing ANCA production by molecular mimicry [15]. Genetic susceptibility to WG is probably polygenic [16]. Whatever the mechanisms predisposing to ANCA generation, there is now good evidence that ANCA are directly involved in the pathogenesis of AAV. MPO-ANCA pathogenicity has been established in animal models [17] and in at least two cases of transplacental transmission in humans resulting in affected neonates [18,19].
Pathology.
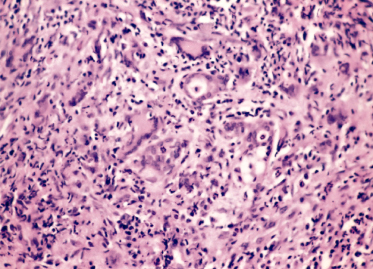
Wegener granulomatosis is a necrotizing granulomatous vasculitis, predominantly affecting small arteries and veins, which are infiltrated with polymorphonuclear leucocytes followed by mononuclear cells. Granulomas are particularly seen in the upper and lower airways (Fig. 167.2), although there may be an absence of the classic histological changes, with the only abnormality being chronic inflammation. Renal lesions are variable; a focal and segmental glomerulonephritis is most commonly found although a diffuse proliferative glomerulonephritis with crescents is seen in those with a rapidly progressive clinical course and marked renal functional decline. Granulomas and evidence of vasculitis are often absent in renal biopsies. Skin biopsies may give supportive evidence for the diagnosis of WG. In a report of WG in a 10-year-old child, the finding of a leucocytoclastic vasculitis with organizing thrombi in the dermis and panniculus in the absence of granulomatous changes was typical of changes that are consistent with but not absolutely diagnostic of WG [20].
Fig. 167.2 Nasal biopsy of WG: granulomatous inflammation with giant cells and generalized destructive acute inflammation involving nasal mucous glands.
Clinical Features.
From a clinical perspective, it may be useful to think of WG as having two forms: a predominantly granulomatous form with mainly localized disease with chronic course, and a florid, acute small vessel vasculitic form characterized by severe pulmonary haemorrhage and/or rapidly progressive glomerulonephritis or other severe vasculitic manifestation (see below) [5]. These two broad presentations may co-exist or present sequentially in individual patients.
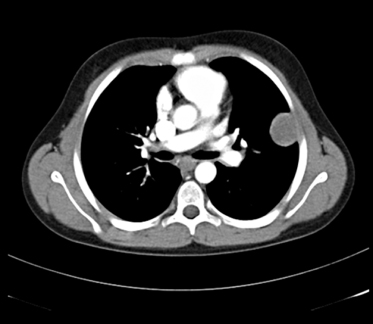
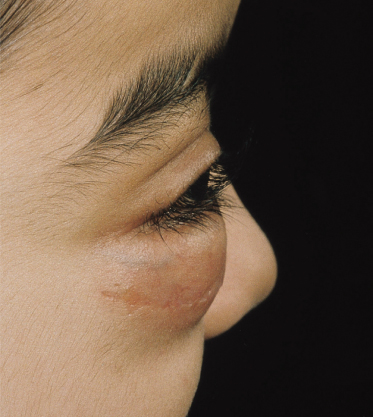
The upper respiratory tract and ears are often affected first [21]. Subacute or chronic otitis may cause mastoiditis; hearing loss can be secondary to serous otitis or sensorineural in origin. Nasal congestion and epistaxis are frequent and ulceration and crusting of the mucosa result from the underlying necrotizing inflammatory process that may lead to septum perforation. Chondritis of the nose results in the typical saddle nose deformity (Fig. 167.3) [22]. Sinuses are often affected, ranging from mucosal thickening to pansinusitis and bone destruction. Oral features include jaw pain, gingival hyperplasia and palatal ulceration. Subglottic stenosis is the most commonly affected area in the tracheobronchial tree in children, affecting up to 48% of children (Fig. 167.4) [11,21]. In a child with stridor, laryngoscopy with biopsies of any lesions in the airway may be helpful diagnostically. Pulmonary involvement may be asymptomatic. Single or multiple nodular masses with or without cavitation may be seen, as can focal or diffuse infiltrates [23] (Fig. 167.5).
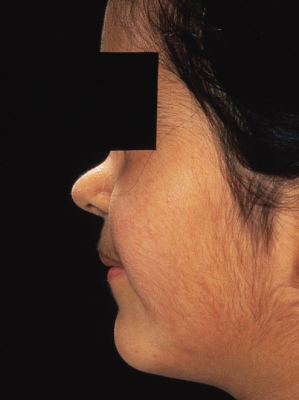
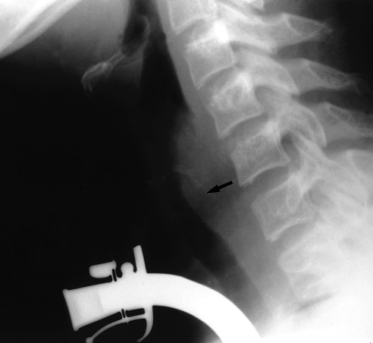
Fig. 167.5 Pulmonary nodule in a child with WG.
Reproduced from Brogan et al. Small vessel vasculitis. Pediatr Nephrol 2010;25:1025–35 with permission from Springer.
Clinical manifestations relating to renal involvement include hypertension, haematuria and/or proteinuria, nephritic and nephrotic syndrome, and in some cases a rapidly progressive nephritis with acute renal failure [24].
Coronary arteritis, myocardial infarction, granulomatous valvulitis of the aortic or mitral valves, pericarditis or pancarditis are all rare complications
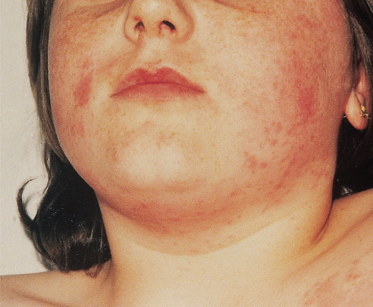
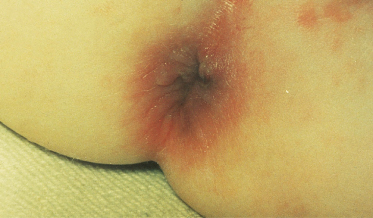
Skin disease may present as erythema, urticarial lesions, petechiae, purpura, ulcerative lesions, pyoderma gangrenosum and necrotic lesions [11,20,25–33] (Fig. 167.6). In both paediatric and adult series, skin disease is reported in approximately 50% [4,11,21]. In one large series of 244 adults, 30 (14%) were found to have skin involvement and detailed clinical, histopathological and cANCA findings were reported [27]. Initial presentation with skin disease is less common, occurring in 8.6–13% of cases [21,25,26]. No single lesion is pathognomonic and lesions must be considered together with other findings. One case report described a child presenting with nodular and necrotic acneiform lesions predominantly on her forehead, which then progressed to ulcers on the arms and buttocks [25] but at presentation, other features of WG were also identified.
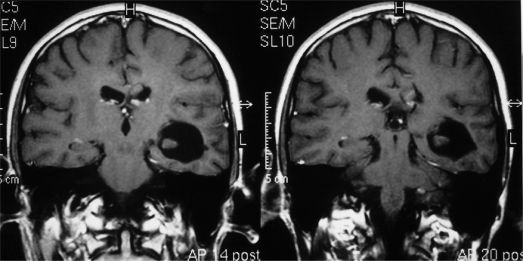
Migratory arthralgias may affect several joints and symmetrical polyarthritis can occur but destructive arthritis is rare. Neurological involvement may result in cranial nerve palsies, mononeuritis multiplex, symmetrical peripheral neuropathy, cerebral infarction and transverse myelitis (Fig. 167.7). Ophthalmological disease causing conjunctivitis, episcleritis, corneoscleral ulcers, uveitis, vasculitis of the retina, optic neuritis and central artery occlusion is documented. Unilateral or bilateral proptosis may be caused by granulomatous pseudo-tumours of the orbit [34,35] (Fig. 167.8). Gastrointestinal symptoms are rare although vasculitic ischaemia may result in bowel perforation, and necrotizing vasculitic involvement of the anus and rectum has been seen (Fig. 167.9).
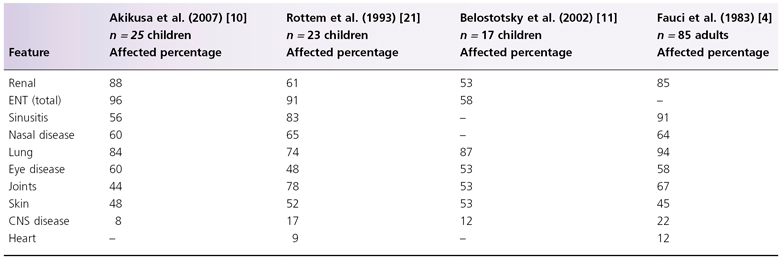
It is known that a limited form of WG also exists, in which the pathological findings are confined to the respiratory tract [36]. Subglottic stenosis is a feature of WG that has been reported in the absence of other clinical markers but in association with ANCA, demonstrating the benefit of a laboratory marker that allows early diagnosis and intervention with appropriate treatment [37]. A comparison of the clinical findings in children and adults is shown in Table 167.1.
Table 167.1 Comparison of features of WG in children and adults [4,10,11,21]
In addition to the haematological and immunological investigations discussed above, radiology can be of diagnostic benefit in WG [38]. Chest radiograph may demonstrate pulmonary infiltrates or discrete nodular and/or cavitating lesions; sinus radiographs may be abnormal and neck views may demonstrate subglottic stenosis [23,28].
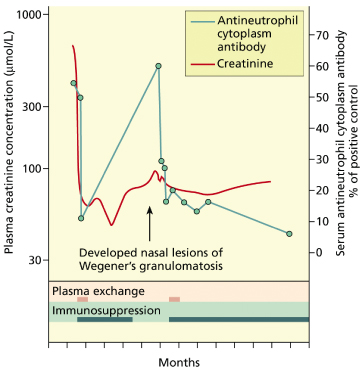
Whilst the diagnostic value of ANCA is without question important, ANCA for the longitudinal monitoring of disease activity is probably unreliable for many patients with WG [13] although it can be used in some for this purpose (Fig 167.10). The reasons for this are not fully understood but are likely to include methodological limitations of ANCA detection, the partial dissociation of ANCA levels and disease activity associated with immunosuppressive therapy, and as yet undefined complexity in the exact role of ANCA in the pathogenesis of WG [39].
Treatment.
Renal morbidity and mortality are a major concern in WG and other AAV, so therapy aimed at preservation of renal function is a recurring theme for the treatment of WG in adults and children [39]. Treatment for paediatric WG is broadly similar to the approach in adults, with corticosteroids, cyclophosphamide (usually 6–10 IV doses at 500–1000 mg/m2 per dose given 3–4 weekly; alternatively given orally at 2 mg/kg/day for 2–3 months) and plasma exchange (particularly for pulmonary capillaritis and/or rapidly progressive glomerulonephritis – ‘pulmonary-renal syndrome’) routinely employed to induce remission [5,39,40]. Intravenous pulsed cyclophosphamide is increasingly favoured over oral continuous cyclophosphamide in adults because of a reduced cumulative dose and less neutropenic sepsis [41] and it is therefore increasingly used to treat children with AAV as well, albeit without good paediatric evidence. This is followed by low-dose corticosteroids and azathioprine (1.5–3 mg/kg/day) to maintain remission [42,43]. Antiplatelet doses of aspirin (1–5 mg/kg once a day) could also be considered empirically on the basis of the increased risk of thrombosis associated with the disease process [44]. Methotrexate may have a role for induction of remission in patients with limited WG [45] but is less commonly used as an induction agent in children with AAV. Co-trimoxazole is commonly added for the treatment of WG particularly in those with upper respiratory tract involvement, serving both as prophylaxis against opportunistic infection and as a possible disease-modifying agent [46]. Recommendations regarding duration of maintenance therapy are based on adult trial data suggesting that the strongest predictor of relapse is withdrawal of therapy, indicating that maintenance therapy should be continued for several years [39]. As a general therapeutic measure, prophylaxis against osteoporosis, gastrointestinal ulceration and infection (bacterial, and fungal) is a standard additional aspect of treatment for AAV [39].
As the use of cyclophosphamide contributes to morbidity and mortality [39,42], with infection playing a prominent role [47], and disease relapses occur in 50% of the patients with AAV as drugs are reduced or withdrawn, newer immunosuppressive agents and immunomodulatory strategies are being explored in both adults and children. Such treatments include mycophenolate mofetil (MMF) and rituximab, which have already been reported to be effective at inducing or maintaining remission in adults with AAV [48,49] and are currently being evaluated in randomized controlled trials (RCTs).
Increasingly, biological therapy is being used to treat children with small vessel vasculitis including AAV as well as ANCA-negative vasculitides. Agents used include rituximab (previously mentioned), anti-tumour necrosis factor (TNF)-α (etanercept, infliximab and adalimumab) and anakinra (recombinant interleukin-1 receptor antagonist) [50]. These therapies are mainly reserved for those who have failed standard treatment or those patients in whom cumulative cyclophosphamide and/or corticosteroid toxicity is of particular concern. Other novel therapies for AAV are reviewed elsewhere [39].
Specialist localized treatment of the upper and lower respiratory tract involvement of WG is beyond the scope of this chapter; the reader is directed to the report of Hoffman et al. [51] and the recent report by White & Shah [52].
Prognosis.
The AAV still carry considerable disease-related morbidity and mortality, particularly due to progressive renal failure or aggressive respiratory involvement, and therapy related complications such as sepsis.
The mortality for paediatric WG in one paediatric series was 12% over a 17-year period of study inclusion [11]. The largest paediatric series of WG reported 40% of cases with chronic renal impairment at 33 months’ follow-up [10] despite therapy. Thus treatment has shifted WG from a disease associated with high mortality in the first year to that of a chronic illness associated with relapsing course and high renal morbidity during paediatric follow-up.
References
1 Radice A, Sinico RA. Antineutrophil cytoplasmic antibodies (ANCA). Autoimmunity 2005;38:93–103.
2 Jennette JC, Falk RJ, Andrassy K et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994;37:187–92.
3 Godman GC, Churg J. Wegener’s granulomatosis: pathology and review of the literature. AMA Arch Pathol 1954;58:533–53.
4 Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983;98:76–85.
5 Brogan P, Eleftheriou D, Dillon M. Small vessel vasculitis. Pediatr Nephrol 2010;25(6): 1025–35.
6 Davies DJ, Moran JE, Niall JF, Ryan GB. Segmental necrotising glomerulonephritis with antineutrophil antibody. Possible arbovirus aetiology? BMJ (Clin Res Ed) 1982;285:606.
7 Rao JK, Weinberger M, Oddone EZ, Allen NB, Landsman P, Feussner JR. The role of antineutrophil cytoplasmic antibody (c-ANCA) testing in the diagnosis of Wegener granulomatosis: a literature review and meta-analysis. Ann Intern Med 1995;123:925–32.
8 Venning MC, Quinn A, Broomhead V, Bird AG. Antibodies directed against neutrophils (C-ANCA and P-ANCA) are of distinct diagnostic value in systemic vasculitis. Q J Med 1990;77:1287–96.
9 Wong SN, Shah V, Dillon MJ. Antineutrophil cytoplasmic antibodies in Wegener’s granulomatosis. Arch Dis Child 1998;79:246–50.
10 Akikusa JD, Schneider R, Harvey EA et al. Clinical features and outcome of pediatric Wegener’s granulomatosis. Arthritis Rheum 2007;57:837–44.
11 Belostotsky VM, Shah V, Dillon MJ. Clinical features in 17 paediatric patients with Wegener granulomatosis. Pediatr Nephrol 2002;17:754–61.
12 Van der Woude FJ, Rasmussen N, Lobatto S et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet 1985;1:425–9.
13 Finkielman JD, Merkel PA, Schroeder D et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007;147:611–19.
14 Harper L, Savage CO. Pathogenesis of ANCA-associated systemic vasculitis. J Pathol 2000;190:349–59.
15 Pendergraft WF III, Preston GA, Shah RR et al. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med 2004;10:72–9.
16 Wieczorek S, Holle JU, Epplen JT. Recent progress in the genetics of Wegener’s granulomatosis and Churg–Strauss syndrome. Curr Opin Rheumatol 2009;1986:49–53.
17 Xiao H, Heeringa P, Hu P et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 2002;110:955–63.
18 Schlieben DJ, Korbet SM, Kimura RE, Schwartz MM, Lewis EJ. Pulmonary-renal syndrome in a newborn with placental transmission of ANCAs. Am J Kidney Dis 2005;45:758–61.
19 Bansal PJ, Tobin MC. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann Allergy Asthma Immunol 2004;93:398–401.
20 Stein SL, Miller LC, Konnikov N. Wegener’s granulomatosis: case report and literature review. Pediatr Dermatol 1998;15:352–6.
21 Rottem M, Fauci AS, Hallahan CW et al. Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr 1993;122:26–31.
22 Orlowski JP, Clough JD, Dyment PG. Wegener’s granulomatosis in the pediatric age group. Pediatrics 1978;61:83–90.
23 Wadsworth DT, Siegel MJ, Day DL. Wegener’s granulomatosis in children: chest radiographic manifestations. Am J Roentgenol 1994;163:901–4.
24 Hall SL, Miller LC, Duggan E, Mauer SM, Beatty EC, Hellerstein S. Wegener granulomatosis in pediatric patients. J Pediatr 1985;106:739–44.
25 Brazzelli V, Vassallo C, Baldini F, Ravelli A, Martini A, Borroni G. Wegener granulomatosis in a child: cutaneous findings as the presenting signs. Pediatr Dermatol 1999;16:277–80.
26 Chyu JY, Hagstrom WJ, Soltani K, Faibisoff B, Whitney DH. Wegener’s granulomatosis in childhood: cutaneous manifestations as the presenting signs. J Am Acad Dermatol 1984;10:341–6.
27 Daoud MS, Gibson LE, DeRemee RA, Specks U, el-Azhary RA, Su WP. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol 1994;31:605–12.
28 Frances C, Du LT, Piette JC et al. Wegener’s granulomatosis. Dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol 1994;130:861–7.
29 Hisler BM, Saltzman L. Cutaneous involvement in Wegener’s granulomatosis. Cutis 1991;48:460–1.
30 Mangold MC, Callen JP. Cutaneous leukocytoclastic vasculitis associated with active Wegener’s granulomatosis. J Am Acad Dermatol 1992;26:579–84.
31 Patten SF, Tomecki KJ. Wegener’s granulomatosis: cutaneous and oral mucosal disease. J Am Acad Dermatol 1993;28:710–18.
32 Spigel GT, Krall RA, Hilal A. Limited Wegener’s granulomatosis. Unusual cutaneous, radiographic, and pathologic manifestations. Cutis 1983;32:41–51.
33 Thomas RH, Payne CM, Black MM. Wegener’s granulomatosis presenting as pyoderma gangrenosum. Clin Exp Dermatol 1982;7:523–7.
34 Haynes BF, Fishman ML, Fauci AS, Wolff SM. The ocular manifestations of Wegener’s granulomatosis. Fifteen years’ experience and review of the literature. Am J Med 1977;63:131–41.
35 Sacks RD, Stock EL, Crawford SE, Greenwald MJ, O’Grady RB. Scleritis and Wegener’s granulomatosis in children. Am J Ophthalmol 1991;111:430–3.
36 Carrington CB, Liebow A. Limited forms of angiitis and granulomatosis of Wegener’s type. Am J Med 1966;41:497–527.
37 Hoare TJ, Rhys Evans PH. Anti-neutrophil cytoplasmic antibody assay in diagnosis of recurrent subglottic stenosis. Lancet 1988;2:1360.
38 Neumann G, Benz-Bohm G, Rister M. Wegener’s granulomatosis in childhood. Review of the literature and case report. Pediatr Radiol 1984;14:267–71.
39 Jayne D. Review article: progress of treatment in ANCA-associated vasculitis. Nephrology 2009;14:42–8.
40 Wright E, Dillon MJ, Tullus K. Childhood vasculitis and plasma exchange. Eur J Pediatr 2007;166:145–51.
41 De Groot K, Adu D, Savage CO. The value of pulse cyclophosphamide in ANCA-associated vasculitis: meta-analysis and critical review. Nephrol Dial Transplant 2001;16:2018–27.
42 Dillon MJ. Vasculitis treatment. New therapeutic approaches. Eur J Pediatr 2006;165:351–7.
43 Jayne D, Rasmussen N, Andrassy K et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349:36–44.
44 Stassen PM, Derks RP, Kallenberg CG, Stegeman CA. Venous thromboembolism in ANCA-associated vasculitis: incidence and risk factors. Rheumatology (Oxford) 2008:47(4):530–4.
45 De Groot K, Rasmussen N, Bacon PA et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005;52:2461–9.
46 Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med 1996;335:16–20.
47 Beimler JH, Andrassy K. Cyclophosphamide treatment in systemic necrotizing vasculitis and lupus nephritis. How long? How much? Pediatr Nephrol 2004;19:949–55.
48 Jones RB, Ferraro AJ, Chaudhry AN et al. A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2009;60:2156–68.
49 Joy MS, Hogan SL, Jennette JC, Falk RJ, Nachman PH. A pilot study using mycophenolate mofetil in relapsing or resistant ANCA small vessel vasculitis. Nephrol Dial Transplant 2005;20:2725–32.
50 Eleftheriou D, Melo M, Marks SD et al. Biologic therapy in primary systemic vasculitis of the young. Rheumatology (Oxford) 2009;48:978–86.
51 Hoffman GS, Thomas-Golbanov CK, Chan J, Akst LM, Eliachar I. Treatment of subglottic stenosis, due to Wegener’s granulomatosis, with intralesional corticosteroids and dilation. J Rheumatol 2003;30:1017–21.
52 White JB, Shah RK. Wegener’s granulomatosis of the pediatric airway: a case demonstrating a conservative management approach. Am J Otolaryngol 2009;30:212–15.
Polyarteritis Nodosa and Microscopic Polyangiitis
Definition.
Classic (macroscopic) polyarteritis nodosa (PAN) predominantly affects medium-sized arteries, and should be differentiated from the small vessel ANCA-associated vasculitis microscopic polyangiitis (MPA), which is now classified separately from PAN [1] but was historically referred to as ‘microscopic PAN’. Thus, for the purposes of this chapter, MPA and PAN will be described separately, although in reality there is a varying degree of overlap between these entities in some patients.
Microscopic Polyangiitis
Definition.
Microscopic polyangiitis (MPA, formerly microscopic polyarteritis) differs from classic PAN by the presence of extensive glomerular involvement, and may be defined as necrotizing vasculitis, with few or no immune deposits, affecting small vessels (i.e. capillaries, venules or arterioles); necrotizing arteritis involving small and medium-sized arteries may be present; necrotizing glomerulonephritis is very common; pulmonary capillaritis often occurs [2]. Clinically, it can be difficult to distinguish from Wegener granulomatosis and often presents with rapidly progressive pauci-immune glomerulonephritis [3] in association with perinuclear antineutrophil cytoplasmic antibody (pANCA, MPO-ANCA) positivity [2].
Renal-limited AAV describes those with rapidly progressive glomerulonephritis, often with ANCA positivity (usually MPO-ANCA) but without other organ involvement, and probably represents a forme fruste MPA [4].
Clinical Features.
The typical clinical manifestations are rapidly progressive glomerulonephritis and alveolar haemorrhage. Other possible symptoms resemble those encountered in polyarteritis nodosa (see below). In adults, 75–80% of patients have pANCA/MPO-ANCA. MPO-ANCA pathogenicity has been established in animal models [5] and in at least two cases of transplacental transmission in humans resulting in affected neonates [6,7]. Renal limited forms are described in children and adults.
Treatment and Prognosis.
The treatment of MPA is similar to that previously described for WG, with corticosteroids and cyclophosphamide used for the induction of remission, followed by maintenance of remission with azathioprine. Plasma exchange should be considered for those with rapidly progressive glomerulonephritis [8,9] and newer agents, including mycophenolate mofetil and rituximab, are also increasingly used in children and adults [10].
For MPA in children, mortality during paediatric follow-up is reportedly 0–14% [11,12]. Two of seven children reported by Peco-Antic et al. [13] progressed to end-stage renal disease; one developed chronic renal failure and four normalized renal function. A selected population of 31 children with necrotizing pauci-immune glomerulonephritis and positive ANCA from Japan reported by Hattori et al. suggested a poor renal prognosis despite therapy [12]. In that series, at 43 months’ follow-up, 29% developed end-stage renal failure, 19.4% had reduced renal function and only 48.4% had normal renal function. Using these data, the authors suggested overall 75% renal survival at 39 months, comparable to the poor renal prognosis of MPA in adults [14].
Polyarteritis Nodosa
Polyarteritis nodosa (PAN) was first described in 1866 by Kussmaul and Maier in the autopsy of a 27-year-old man who had proteinuria, myalgia, neuritis and abdominal pain. It is a disease of small and medium-sized arteries with aneurysmal dilation, especially at arterial branching points. Although it is rare, PAN does occur in childhood [15–18]. There is a debate about the overlap of infantile polyarteritis with Kawasaki disease [19]. Some of the early reports of infantile polyarteritis give classic descriptions of Kawasaki disease [20,21]. PAN may present as multisystem disease but there is also a group of patients in whom the cutaneous manifestations predominate and in whom the prognosis is considerably better [22–24].
In adults from Europe and the United States, the estimated annual incidence of PAN is 2–9/million [25]. Although comparatively rare in childhood, it is the most common form of systemic vasculitis after Henoch–Schönlein purpura and Kawasaki disease [26,27]. However, the data are difficult to interpret since the incidence in various reports differs widely dependent on which criteria are used to classify patients into the PAN category. New criteria for the classification of PAN in children have recently been described (Box 167.1). Sensitivity and specificity of these new criteria were 73% and 100% respectively [28].
Box 167.1 New Criteria for the Classification PAN in Children
Histopathological evidence of necrotizing vasculitis in medium or small arteries or angiographic abnormality (aneurysm, stenosis or occlusion) as a mandatory criterion plus one of the following five:
- skin involvement (see below)
- myalgia or muscle tenderness
- hypertension
- peripheral neuropathy
- renal involvement
Aetiology and Pathogenesis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


