History.
The term ‘purpura fulminans’ was first introduced by Henoch in 1887 [3]. Since then an extensive literature on the disorder has been published, containing clear descriptions of the clinical features of the disorder and its histopathology [1-4]. The term is now accepted as applying to patients with a devastating illness associated with extensive areas of purpura [2,3]. Despite the large number of cases reported in the literature, the cause of the disorder has remained unclear and a confusing, and often conflicting, array of different theories have been presented on its aetiology and pathogenesis. A large number of different processes have been reported to initiate purpura fulminans, including bacterial and viral infection [4], autoimmune diseases [5], vasculitis and a number of drugs [6]. Several disease mechanisms have been proposed to explain the central feature of the disorder – the widespread thrombosis within dermal blood vessels. These include endothelial injury, primary activation of coagulation pathways, impaired anticoagulation mechanisms and activation of platelets [2,4,6].
Not surprisingly in view of the confusion surrounding the aetiology and pathogenesis, the literature also contains conflicting recommendations as to treatment. Faced with a progressive and life-threatening illness of unclear aetiology, clinicians have utilized a wide variety of experimental or unproven treatments for the disorder. These include clotting factor replacement [7], anticoagulation [8], antiplatelet agents, glucocorticosteroids and immunosuppression [4], plasmapharesis or whole blood exchange [9], hyperbaric oxygen [10] and fibrinolytic agents [11].
Recognition that congenital deficiencies of protein C or S are associated with purpura fulminans [12,13] has highlighted the importance of the protein C pathway in the aetiology of the disorder. More recently, reports of purpura fulminans associated with acquired deficiency of protein C or S, either occurring during fulminant sepsis [14,15] or resulting from autoimmune processes [16], have highlighted the importance of primary thrombotic processes in initiation of the disorder. The reports published in the past few years have not only provided a clearer understanding of the pathogenesis of the disorder, but also increasingly allow purpura fulminans to be understood as a syndrome that can result from several distinct disease processes.
Classification of Purpura Fulminans Based on Aetiology and Pathogenesis.
As with many other clinical disorders that were initially defined simply by description of the major clinical and histopathological features, it is now apparent that purpura fulminans is not a single disease but a common clinical and histopathological manifestation of a number of distinct disease processes. Thus the major clinical feature (extensive purpura) is the result of a single histopathological process [17,18] (dermal vascular thrombosis and haemorrhagic infarction of the surrounding tissues). These clinical and histopathological features may be caused by a wide range of different disease processes involving the blood vessel wall, platelets or prothrombotic or antithrombotic pathways. Effective treatment of individual patients depends on identification of the underlying aetiological and pathophysiological mechanism, and the introduction of specific treatments to correct the disordered physiological process.
In this chapter a classification of purpura fulminans is proposed which enables patients presenting with purpura fulminans to be classified into one of eight groups on the basis of clinical and epidemiological criteria and laboratory findings (Table 162.1). Each of these groups has a distinct aetiology and pathogenesis. Treatment recommendations are based on the current understanding of the pathophysiological mechanisms.
Table 162.1 Classification of purpura fulminans based on aetiology and pathogenesis
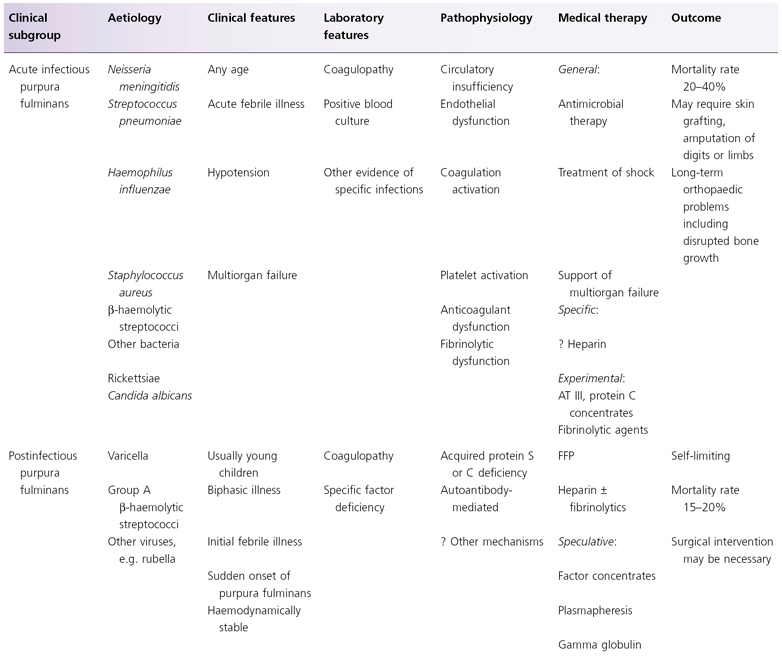


APTT, activated partial thromboplastin time; AT III, antithrombin III; FFP, fresh-frozen plasma; PT, plasma thromboplastin; SLE, systemic lupus erythematosus.
Acute Infectious Purpura Fulminans
Purpura fulminans occurring in the context of acute bacterial sepsis is the most common form of the disorder. A variety of different bacteria have been associated with purpura fulminans, including Staphylococcus aureus [19], groups A and B β-haemolytic streptococci [20,21], Streptococcus pneumoniae [22], Haemophilus influenzae [23], H. aegyptius [24] and Pseudomonas aeruginosa [25]. Although purpura fulminans can be seen as an occasional complication of any of these bacterial infections, it occurs so commonly in the context of Neisseria meningitidis infection that its presence is considered a cardinal feature of meningococcal septicaemia [11,26,27]. Purpura fulminans associated with acute bacterial sepsis occurs in all age groups, but is most common in children. Patients with purpura fulminans associated with acute bacterial infection such as meningococcaemia invariably present with the features of systemic infection, and the majority will have evidence of shock [26,28-30]. The presenting features are often high fever, shivering, muscle aches, vomiting and abdominal pain. A particularly common presenting feature of children with severe purpura fulminans in the context of sepsis is intense limb pain, which may be an early sign of venous thrombosis. As the disease progresses, confusion, impaired consciousness, laboured breathing and other signs of systemic underperfusion become more apparent. Signs of shock are usually present including tachycardia, prolonged capillary refill, a wide gap between central and peripheral temperature, oliguria and elevated respiratory rate. Hypotension ultimately occurs once compensatory vasoconstrictive mechanisms are unable to maintain blood pressure in the face of severe shock. The disorder is frequently associated with multiorgan failure, but with recent advances in early recognition and aggressive treatment of the disease, childhood mortality rates of 20–40% have been reduced to below 5% in countries where modern paediatric intensive care is available [26,27,31,32].
The pathophysiology of sepsis associated with purpura fulminans is complex (see Chapter 55). Bacteria or their toxins (principally lipo-oligosaccharide in the case of Gram-negative organisms) trigger an intense inflammatory process, with activation of neutrophils, macrophages, complement and the clotting cascade [29,33]. Endothelial injury mediated by bacterial toxins directly or secondary to host inflammatory factors, such as tumour necrosis factor (TNF), interleukin 1 (IL-1), reactive oxygen intermediates or proteolytic enzymes, results in disruption of the endothelium and loss of antithrombotic mechanisms [33]. Endotoxin induces upregulation of adhesion molecules on the endothelial surface, which facilitate the attachment of neutrophils to the endothelial surface [33,34]. Upregulation of endothelial procoagulant activities, including tissue factor, occurs [35]. Neutrophils, activated by endotoxin, induce loss of the anticoagulant glycosaminoglycans, heparin sulphate and chondroitin sulphate from the endothelial surface [36], downregulation of prostacyclin production [37] and a defect in the activation of antithrombin by the endothelium [38].
The normal regulatory systems preventing uncontrolled coagulation are disturbed. Acquired deficiencies of tissue factor pathway inhibitor [39], antithrombin and proteins C and S [14,15,18,40,41] are caused by the profound capillary leak, together with consumption by the thrombotic processes. Reduced thrombomodulin and endothelial protein C receptor (EPCR) expression on the endothelium cause a defect of protein C activation in the dermal vessels [18]. The fibrinolytic system is also impaired, caused by elevation of plasminogen activator inhibitor 1 (PAI-1), the physiological inhibitor of tissue plasminogen activator (t-PA). In sepsis, elevated PAI-1 has been correlated with the development of shock, renal impairment and mortality [42]. In meningococcaemia, an increase in PAI-1 has been associated with death [43]. Variation in the PAI-1 gene does not affect the probability of an individual contracting meningococcal disease, but instead influences whether that person will develop septic shock or die from the disease [44,45].
The intravascular thrombosis seen in infectious purpura fulminans is therefore due to a combination of sluggish vascular circulation due to shock and platelet degranulation leading to local vasoconstriction [33], together with upregulation of procoagulant pathways and downregulation of anticoagulation regulatory pathways [33,46].
Postinfectious Purpura Fulminans
In contrast to purpura fulminans occurring as a complication of acute bacterial sepsis, ‘idiopathic’ or ‘postinfectious purpura fulminans’ characteristically occurs 1–3 weeks after an acute infectious process [4]. The disorder is more common in young children, and varicella and streptococcal infections are the most common antecedents [4], although a variety of other childhood illnesses have been reported to precede the disorder [1,4,16]. The disorder follows a biphasic course. After appearing to recover from an otherwise uncomplicated childhood illness, affected patients suddenly develop extensive areas of purpura, principally affecting the buttocks and lower limbs (Fig. 162.1b). Patients are usually afebrile and, except for areas of skin infarction or peripheral gangrene, are well perfused and normotensive. The disease may progress rapidly to cause extensive areas of skin necrosis and gangrene of the limbs or digits. Thromboembolic complications of internal organs may subsequently occur. Thrombosis of large vessels may lead to pulmonary emboli, and embolization or thrombosis within the kidneys, brain, heart or other organs [16,47].
Affected patients have laboratory evidence of disseminated intravascular coagulation, with prolongation of prothrombin time, partial thromboplastin time and thrombin time, hypofibrinogenaemia and elevation of fibrin degradation products.
A wide range of different theories on the aetiology of the disorder have been presented. Although it is possible that the purpura fulminans following varicella may be mediated by a different process from that occurring in the context of streptococcal infections or other viral infections [4,6], there is now clear evidence that an acquired deficiency of protein S [47,48], induced by autoantibodies against protein S, is a consistent feature of the disorder [16]. In 1993, d’Angelo et al. reported a single case of thromboembolic disease occurring in a child who had recently had varicella [49]. The patient had an acquired deficiency of protein S caused by autoantibodies directed against protein S. Although their patient did not have purpura fulminans, or any cutaneous manifestations of thrombosis, their report led the authors to search for the presence of autoantibodies in similar patients with postvaricella purpura fulminans. In five consecutive children presenting with purpura fulminans following varicella or other viral infections, the authors documented severe acquired protein S deficiency caused by immunoglobulin G (IgG) or IgM autoantibodies directed against protein S, and this now appears to be the common mechanism underlying postinfectious purpura fulminans [16].
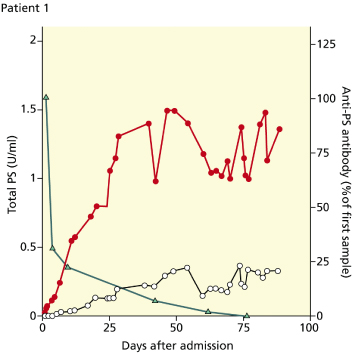
Varicella or other bacterial or viral infections may trigger the production of an autoantibody, which cross-reacts with the protein S. As no known varicella proteins exist with similar structure to that of protein S, it is likely that the antibody is directed against a neo-antigen exposed during the varicella infection. The antibodies appear to increase the clearance of protein S, perhaps by rapid removal of the protein S antibody complex, through binding to Fc receptors on the reticuloendothelial system. The enhanced clearance of protein S not only explains the low levels of protein S present at the time of admission, but also explains the difficulties which may be found in restoring protein S levels to normal even following the infusion of large volumes of plasma. Levels of the antiprotein S autoantibodies decline spontaneously within 1–6 weeks after the onset of the disease [16] (Fig. 162.2). As IgG responses would normally be expected to be long lasting, the rapid disappearance of the antibody from the circulation may suggest involvement of an anti-idiotype response. Anticardiolipin antibodies are present in some of the patients with postvaricella purpura fulminans, including those with antiprotein S antibodies. Low titres of IgM and IgG anticardiolipin antibodies are detected in some of the patients with antiprotein S antibodies and may be implicated in the pathogenesis but it is not yet clear whether this represents a cross-reaction or the production of two distinct sorts of cross-reacting antibodies [16,50]. It is also possible that autoantibodies directed against protein C may cause a similar picture in some cases.
Fig. 162.2 Postinfectious purpura fulminans following varicella. The time courses of changes in protein S level are shown following admission of a child with purpura fulminans. On admission both free protein S ( ) and total protein S (
) and total protein S ( ) levels were undetectable. Levels of antiprotein S antibodies were markedly increased (
) levels were undetectable. Levels of antiprotein S antibodies were markedly increased ( ). Over the next 4 weeks, antiprotein S antibody levels progressively declined, with a concurrent rise in both free and total protein S.
). Over the next 4 weeks, antiprotein S antibody levels progressively declined, with a concurrent rise in both free and total protein S.
Congenital Purpura Fulminans Due to Defects in the Protein C or S Pathway
Children with congenital protein C and S deficiency may present in the neonatal period with thrombosis of major organs including the brain with or without the cutaneous manifestations of purpura fulminans [12,51]. Severely affected patients have either homozygous protein C or S deficiency, or are functionally homozygous due to compound heterozygous states. The cutaneous features of purpura fulminans may occur spontaneously either in the neonatal period or within the first months of life. In some cases, cutaneous or internal organ thrombosis occurs spontaneously but in other cases, a precipitating infectious or inflammatory insult appears to trigger the disease. Unlike those patients with acute infectious purpura fulminans, those with congenital protein C or S deficiency are often haemodynamically stable and afebrile at the time of presentation unless an infection has precipitated the onset of purpura. A family history of thromboembolism is common as the heterozygous carriers of the disorder are at increased risk of thromboembolism. Any child presenting in the neonatal period or early months of life with purpura fulminans or thromboembolism should be suspected of having protein C or S deficiency and should be investigated appropriately [12,13,51–53].
Acquired Protein C or S Deficiency Due to Drugs or Specific Diseases
Purpura fulminans has increasingly been recognized in patients treated with drugs such as coumarin derivatives, which suppress production of protein C and S, in addition to the production of the vitamin K-dependent clotting factors [6]. Patients heterozygous for deficiency of protein C or S appear to be at risk [54–56]. Acquired protein C and S deficiency may also occur in patients with cholestatic hepatic disease [57], the nephrotic syndrome, peritoneal dialysis [58] and bone marrow transplantation [59]. The clinical features in this subgroup include the use of drugs which affect protein C or S production or the presence of underlying predisposing diseases. In the case of coumarin drug usage, patients developing purpura fulminans appear to have a more rapid decline in levels of protein C and S than the desired depletion of the vitamin K-dependent procoagulant factors. Patients heterozygous for protein C or S deficiency are at risk of this complication. Patients with the nephrotic syndrome have increased urinary clearance of protein C and S as part of the generalized proteinuria. Impaired production of protein C and S or increased clearance underlies the association of depletion of these proteins with hepatic disease or dialysis.
Purpura Fulminans Associated with systemic vasculitic disorders and the Antiphospholipid Antibody Syndrome
Purpura fulminans may occur in patients with systemic autoimmune disease such as systemic lupus erythematosus (SLE) [60], polyarteritis nodosa or Henoch–Schönlein purpura [61]. It may also occur as a component of the antiphospholipid antibody syndrome [62,63] (Fig. 162.3).
Fig. 162.3 Purpura fulminans and peripheral gangrene in a child with systemic lupus erythematosus and antiphospholipid antibodies.
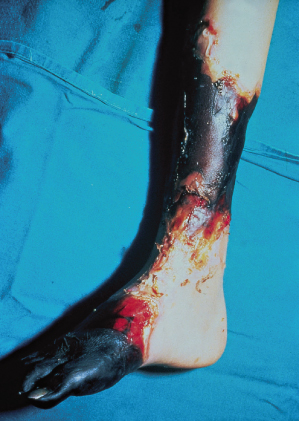
The clinical features of patients presenting with purpura fulminans in the context of these systemic disorders are usually dominated by those of the systemic illness. Patients with SLE or polyarteritis nodosa may have a prolonged febrile illness with evidence of multiorgan involvement including arthritis, nephritis, central nervous system disease or pneumonitis [64,65]. Occasionally, patients with antiphospholipid antibodies may have no underlying systemic illness and may present acutely with either purpura fulminans or major organ thrombosis [62,63].
Laboratory features include an acute inflammatory response, with elevation of the erythrocyte sedimentation rate. Neutrophil leucocytosis and elevation of C-reactive protein are common in the acute vasculitides, whereas paradoxical depression of C-reactive protein, neutropenia and thrombocytopenia may occur in SLE. Antiphospholipid antibodies may occur in association with other markers of SLE, including anti-DNA antibodies, antibodies to extractable nuclear antigens, and low C3 and C4. A number of antibody-mediated mechanisms initiating thrombosis have been identified in patients with antiphospholipid antibodies, including inhibition of protein C activation by thrombomodulin, inhibition of the anticoagulant action of activated protein C, and interference with antithrombin 3 binding to endothelial glycosaminoglycans [62,63]. Platelet activation may also occur. Non-antibody mediated mechanisms may also be involved, including acute vasculitic damage to the vessel wall mediated by neutrophils and lymphocytes or other inflammatory cells. Patients with systemic vasculitis may have evidence of antineutrophil cytoplasmic antibodies.
Platelet-Mediated Purpura Fulminans Occurring During Heparin Therapy
Heparin-induced skin necrosis, caused by antibody-mediated platelet aggregation, occurs primarily after subcutaneous administration of heparin, usually at the injection site [66,67]. Platelet-mediated mechanisms have also been proposed to explain purpura fulminans occurring in the course of thrombotic thrombocytopenic purpura (TTP) [68] or paroxysmal nocturnal haemoglobinuria [69].
Diagnosis of platelet-mediated purpura fulminans is usually suggested by its occurrence in the context of heparin treatment or in patients with TTP. The disease is usually self-limiting once heparin therapy is discontinued but treatment with alternative anticoagulant agents such as coumarin derivatives, or the use of platelet inhibitors such as non-steroidal anti-inflammatory agents or prostacyclin, may be required in severe cases.
Purpura Fulminans Following Bites or Envenomation
Purpura fulminans may occur after a number of toxic insults including snake bites or spider bites (see Chapter 73). Activation of coagulation and endothelial injury appear to be the underlying mechanisms. The purpura is usually present locally around the site of the toxin inoculation but, in severe cases, extensive areas of purpura may be apparent.
Pathology.
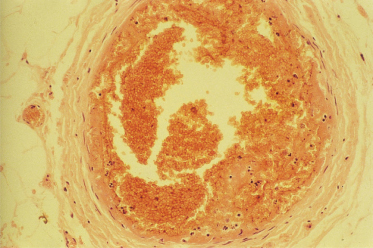
Although purpura fulminans may be caused by a variety of distinct disease processes, the histopathological findings are common to all patients with the disorder. The hallmark of the disease is thrombotic occlusion of dermal vessels [2,4,6,17]. In mild cases the process is confined to the dermal capillaries and venules. In more severe cases thrombosis extends into the deeper tissues involving larger vessels and, in the most severely affected patients, thrombotic occlusion of the major veins draining entire limbs may also occur (Fig. 162.4). Veins and small venules are distended and occluded by fibrin thrombi and large aggregates of red cells. Haemorrhage into the surrounding tissues occurs, resulting in oedema and the appearance of extravascular red blood cells [17].
Fig. 162.4 Histology of purpura fulminans. Thrombosis within the saphenous vein is visible in a child who required below-knee amputation following meningococcal sepsis. The lumen of the vein is occluded by fibrin/thrombus. No inflammatory infiltrate was visible in the vessel wall.
The intravascular thrombosis affecting capillaries, veins and venules frequently occurs without any evidence of underlying vasculitis or inflammatory cell-induced disruption of the vessel wall. This is particularly true of postinfectious purpura fulminans, which is primarily a thrombotic process. In contrast, in patients with purpura fulminans associated with meningococcal sepsis or in vasculitis-associated purpura fulminans, there may be evidence of vasculitis surrounding areas of venous thrombosis [2,18]. Even in patients with purpura fulminans associated with meningococcaemia, venous thrombosis may occur without any evidence of inflammatory cell infiltrate into the vessel wall [26]. Areas of vasculitis may be interspersed between areas of thrombosis without evidence of underlying vessel wall inflammation.
Clinical Features.
The major presenting symptoms and clinical signs in purpura fulminans differ amongst clinical subgroups as described in Table 162.1
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


