Lymph nodes (90%)
Eyes (30%)
Eyes (65%)
TNF, tumour necrosis factor.
Classic Sarcoidosis
Sarcoidosis is a multisystem granulomatous disease of unknown aetiology. It is characterized by noncaseating granulomas that can affect almost any organ, and the clinical manifestations include a broad spectrum of cutaneous findings [2].
Sarcoidosis has a peak incidence in the third and fourth decades of life and is relatively uncommon in children. In a recent Danish study based upon a national registry of hospital discharge diagnoses, the incidence of sarcoidosis in children ≤15 years of age was found to be 0.3 per 100,000 person-years, compared to an overall incidence of 7–10 per 100,000 person-years [4,5]. Classic sarcoidosis occurs more often in adolescents than in younger children, and there is no sex predilection in these age groups [4,6]. In the USA, childhood sarcoidosis is most frequent in the south-eastern and south central regions, with a higher incidence among African-Americans [1]. Outside of the USA, it has been described primarily in children from England, Japan and Scandinavia.
History.
The first report of cutaneous sarcoidosis was published in 1877 by Hutchinson [7], an English dermatologist, ophthalmologist and surgeon. In 1889, the French dermatologist Besnier [8] coined the term ‘lupus pernio’ for violaceous swellings of the nose and fingers, and 3 years later his colleague Tenneson observed that this condition was characterized histologically by epithelioid and giant cells. In 1899, the Norwegian dermatologist Boeck [9] introduced the term ‘multiple benign sarkoid’ for the disorder based upon a patient with multiple skin nodules for which histological evaluation showed giant cells reminiscent of a sarcoma. The multisystem nature of the disease was subsequently recognized, and by the 1930s sarcoidosis was internationally accepted as a distinct clinicopathologic entity [10]. In 1941, the Norwegian dermatologist Kveim observed that intradermal injection of homogenized human sarcoidal tissue led to formation of cutaneous granulomas in patients with sarcoidosis, and this was historically utilized as a diagnostic technique (Kveim test) [11].
Pathogenesis.
It is believed that sarcoidosis results from exposure of genetically susceptible hosts to environmental, occupational or infectious agents that trigger an exaggerated cellular immune response and lead to granuloma formation [12,13]. Despite intensive study, a consistent inciting antigen for sarcoidosis has not been identified. Environmental associations have been suggested by seasonal clustering of onset, and an aetiological role of various chemicals and metals such as aluminum, zirconium and beryllium (which is linked to a sarcoidosis-like granulomatous disorder) has been proposed [14]. Possible occupational risks for sarcoidosis include firefighting, metalworking and employment on navy aircraft carriers [15,16]. Infectious agents such as mycobacteria, Proprionibacterium acnes, fungi and viruses (e.g. Epstein–Barr virus, cytomegalovirus, human herpes virus 8 and human T-lymphotropic virus 1) have been investigated, but a routine role in the pathogenesis of sarcoidosis has not been established for any of these organisms [14,17–20]. For example, Mycobacterium tuberculosis DNA has been found in tissue samples from some children with sarcoidosis and mycobacterial catalase-peroxidase protein (mKatG) has been identified in sarcoidal granulomas [21,22]. Treatment with interferons, particularly in individuals with hepatitis C viral infection, has also been noted to trigger the onset of sarcoidosis [23]. Patients with sarcoidosis have an increased likelihood of developing autoimmune conditions such as systemic lupus erythematosus and autoimmune thyroid disease, suggesting a possible role of autoantigens as well as external antigens.
Twin studies and observations of familial clustering of sarcoidosis indicate a genetic susceptibility to this condition [24–27]. Human leucocyte antigen (HLA) class II variants that have been associated with sarcoidosis include HLA-DQB1 and HLA-DRB1 alleles [24]. HLA-DRB1*0301 and a particular haplotype of the C–C chemokine receptor 2 (CCR2) gene have each been independently linked to acute sarcoidosis with a favourable prognosis [2,24,28]. Recently, an increased risk of sarcoidosis has also been found in individuals with polymorphisms in the butyrophilin-like 2 (BTNL2) gene (located near the HLA genes on chromosome 6) or annexin A11 (ANXA11) gene and in those with a particular IFNA haplotype [29–31].
The inflammation of sarcoidosis is thought to begin when macrophages present an antigen to CD4+ T helper cells within an affected organ. Studies of T-cell receptor gene expression in sarcoidosis patients show oligoclonal collections of CD4+ T cells, consistent with a major histocompatibility complex (MHC)-restricted antigen-driven process. An exaggerated Th1-type immune response results in activation and proliferation of T cells and macrophages, which leads to the formation of granulomas [32]. There is increased production of Th1 cytokines including interferon-γ, interleukin-2 (IL-2) and IL-12. Recruitment of macrophages into granulomas is stimulated by chemokines as well as cytokines such as tumour necrosis factor-α (TNF-α) [32,33]. Recent studies suggest that the apparent paradox of peripheral anergy in patients with sarcoidosis may reflect expansion of regulatory T cells (CD4+, CD25bright) as well as compartmentalization of T cells in peripheral tissues [34]. If granulomatous inflammation persists, presumably because of failure in clearance of the inciting agent or development of reactivity to autoantigens, parenchymal cells and tissue architecture are disrupted, eventuating in progressive fibrosis.
Pathology.
The histopathological hallmark of sarcoidosis is non-caseating granulomas – well-circumscribed collections of tightly packed epithelioid macrophages with minimal or no necrosis [2]. The granulomas are typically associated with only a sparse lymphocytic infiltrate (‘naked’ tubercles) and often contain Langhans-type giant cells within which asteroid bodies (stellate eosinophilic structures representing engulfed collagen) and Schaumann bodies (lamellated basophilic structures representing calcified degenerating lysosomes) may be found [35]. Special stains for organisms (e.g. mycobacteria and fungi) are negative. In patients with systemic sarcoidosis, polarization reveals foreign material in up to 20% of skin biopsy specimens that demonstrate granulomatous involvement; this is most often evident in samples from the elbow or knee, and it is thought to reflect induction of granuloma formation by inoculation of foreign matter and associated trauma (akin to sarcoidosis arising within a tattoo or scar) in predisposed individuals [36].
Clinical Features.
Sarcoidosis is a multisystem disease that can have a wide variety of cutaneous and extracutaneous manifestations. In paediatric and adolescent patients, sarcoidosis most often affects the lungs (up to 90%), lymph nodes (90%) and eyes (30%) as well as the skin (30%) [4,18,37]. In addition, affected children frequently present with constitutional symptoms such as fevers, weight loss and fatigue [4,18,38].
Cutaneous Manifestations
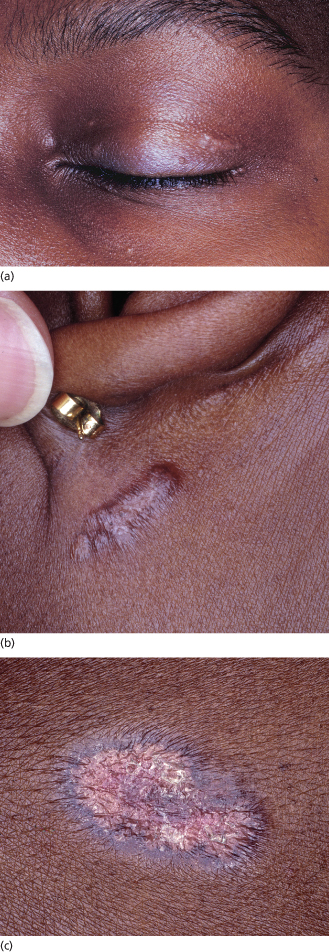
The spectrum of cutaneous findings in patients with sarcoidosis is broad and encompasses both ‘specific’ lesions characterized histologically by non-caseating granulomas and ‘non-specific’ reactive lesions. Cutaneous sarcoidosis may be the initial or only manifestation of the disorder. The most common skin lesions with specific histology are red–brown non-scaly dermal papules and plaques that favour the face (especially the nose, lips and eyelids) (Fig. 158.1), areas of trauma and pre-existing scars. However, the colour can range from yellow–brown to pink to violaceous, epidermal change occasionally leads to plaques with a psoriasiform appearance, and any mucocutaneous site (including the oral mucosa) may be affected [39–42]. Diascopy of erythematous lesions often reveals an ‘apple jelly’ (yellow–brown) colour, which is easiest to appreciate in lightly pigmented skin. The term angiolupoid sarcoidosis has been used for skin lesions with prominent telangiectasias.
Fig. 158.1 Papules and plaques on the eyelid (a), posterior auricular area (b) and back (c) in an adolescent with sarcoidosis. The colour ranges from pink to red-brown to violaceous. Note the annular shape and scale in (c).
Lupus pernio is characterized by indurated violaceous papulonodules and plaques located primarily on the nose, cheeks, ears and hands. This cutaneous sarcoidosis variant, which is rare in children, tends to be persistent and may result in scarring and disfigurement. Lupus pernio is associated with chronic involvement of the respiratory tract (upper and lower) and cystic bone lesions of the phalanges [43]. Other specific cutaneous manifestations of sarcoidosis include subcutaneous nodules (Darier–Roussy sarcoidosis), ichthyosiform eruptions, hypopigmented lesions, ulcers and scarring alopecia [39,41,44,45].
Erythema nodosum is a common non-specific cutaneous manifestation of sarcoidosis which presents with tender erythematous subcutaneous nodules favouring the shins (see Chapter 77). It occurs in up to one-third of children with sarcoidosis and portends a favourable prognosis [4,15]. Erythema nodosum typically develops in the setting of acute disease, often in association with fever, hilar adenopathy, uveitis and polyarthralgias/polyarthritis (Löfgren syndrome) [2].
Extracutaneous Manifestations
The lung and lymph nodes are the most commonly affected organ systems in children (as well as adults) with classic sarcoidosis [2,4]. Patients may present with a dry cough, exertional dyspnoea and chest discomfort [18,46]. Chest radiographs usually reveal bilateral hilar lymphadenopathy (approximately 80% of patients) and/or parenchymal infiltrates (approximately 20% of patients; favour upper and mid lung zones) [1,4,6]. High-resolution computed tomographic (CT) scans of the chest are more sensitive than radiographs in identifying parenchymal and nodal disease and can help to better delineate areas of inflammation and (uncommonly in children) fibrosis. In addition to hilar involvement, almost half of children with sarcoidosis have evidence of peripheral lymphadenopathy [4]. When pulmonary function testing is performed, nearly half of children with active sarcoidosis have evidence of restrictive or, less often, obstructive lung disease [1,4,37,46]. Although bronchoalveolar lavage (BAL) can detect early alveolitis, the presence of lymphocytosis in BAL samples from children with sarcoidosis has not been shown to correlate with disease activity, treatment response or prognosis [47].
Approximately one-third of children with sarcoidosis have ocular involvement, symptoms of which may include eye pain and redness, blurry vision and photophobia [4,48,49]. Anterior uveitis (inflammation of the iris and/or ciliary body) is the most common manifestation, and it can be granulomatous (characterized by large ‘mutton fat’ keratic precipitates or iris nodules) or non-granulomatous [1]. Conjunctival granulomas represent another frequent ocular finding and present as small yellowish nodules [48]. The spectrum of ocular features also includes posterior uveitis (inflammation of the retina or choroid ), intermediate uveitis (inflammation of the vitreous), keratitis, glaucoma and inflammation of the lacrimal glands [4,48].
The granulomatous inflammation of sarcoidosis can affect virtually any organ, including the liver, spleen, bones, muscles, central nervous system, salivary glands, gastrointestinal tract, heart and kidneys [18]. Although arthritis is a major feature of Blau syndrome/EOS (see below), it occurs in only 5% of children with classic sarcoidosis [4]. Neurosarcoidosis in prepubertal children most often presents with seizures and is associated with poor prognosis; additional manifestations include headaches, hypothalamic dysfunction (e.g. diabetes insipidus) and (usually in adolescents) cranial nerve palsies [6,50]. Heerfordt syndrome (uveoparotid fever) refers to the combination of uveitis, parotitis, cranial nerve palsies (typically of the facial nerve) and fever. Cardiac involvement related to granulomatous infiltration may result in arrhythmias or cardiomyopathy [40,51,52]. Renal disease occasionally develops in children with sarcoidosis and is more often secondary to hypercalcaemia and hypercalciuria (resulting from 1,25-dihydroxyvitamin D3 production via 1-α hydroxylase activity within granulomas) than to granulomatous inflammation within the kidneys [1,18,38].
Laboratory Findings
More than half of children with sarcoidosis have an elevated serum angiotensin-converting enzyme (ACE) level [4,53], which is thought to reflect ACE production by the granulomas and can be followed as a marker of disease activity. Of note, normal ACE levels are approximately 50% higher in children than in adults. Other common laboratory abnormalities in children with sarcoidosis include an elevated erythrocyte sedimentation rate, leucopenia (in particular lymphopenia), hypergammaglobulinaemia and hypercalcaemia [4,18,38]. Skin testing may demonstrate impaired delayed-type hypersensitivity reactions [1,18]. In addition to organ-specific radiographical studies, positron emission tomography may be useful in assessing the sites and extent of internal involvement.
Prognosis.
The prognosis of classic sarcoidosis in children and adolescents is generally good, with improvement or complete disease resolution in most patients [6,38]. In a recent long-term study (median follow-up 15 years) of 46 Danish children with sarcoidosis, 80% of the patients recovered completely without functional impairment while the remainder either had persistent disease (13%) or died of neurosarcoidosis or a secondary malignancy (7%) [6]. Clinical resolution occurred a median of 8 months after diagnosis (range 6 months to 6 years), and prognosis was not related to age at disease onset.
Differential Diagnosis.
Diagnosis of sarcoidosis requires a clinical presentation consistent with the disorder, histological documentation of non-caseating granulomas and exclusion of other granulomatous disorders (infectious and non-infectious). Because of the great variety of clinical presentations of cutaneous sarcoidosis, it can mimic a wide range of other skin disorders and biopsy is frequently helpful in narrowing the differential diagnosis [14,39]. Perioral/periorificial dermatitis, a common paediatric condition, can have clinical and histological findings that closely resemble those of sarcoidosis. Foreign body reactions represent another frequent histological mimic and (as noted above) the diagnoses of foreign body granuloma and sarcoidosis are not mutually exclusive. A widespread distribution of small papules with sarcoidal histology in an infant or young child should prompt consideration of Blau syndrome/EOS (see below), even in the absence of arthritis, uveitis and a positive family history. Additional conditions with histological appearances similar to that of sarcoidosis include cutaneous Crohn disease (favouring the groin and lower extremities), granulomatous cheilitis and sarcoidal reactions to an underlying lymphoma. When mycobacterial (e.g. lupus vulgaris) or fungal infection is a clinical possibility, it is important to perform tissue culture as well as special stains for these organisms. Children with granulomatous dermatitis and a history of recurrent, severe or atypical infections should be evaluated for a primary immunodeficiency disorder (see Chapter 177) [54].
Treatment.
The choice of therapy for children with sarcoidosis depends upon the organs involved and disease severity. Corticosteroids represent the mainstay of sarcoidosis treatment [1,55–57]. Intralesional or high-potency topical corticosteroids can be effective for limited skin involvement, while oral prednisone/prednisolone (typically ∼1 mg/kg/day for 1–3 months then tapering and completion of a 6–12-month course) may be indicated for extensive or disfiguring skin lesions as well as for progressive or function-threatening systemic disease. Antimalarial agents (hydroxychloroquine ≤6.5 mg/kg/day or chloroquine ≤3.5 mg/kg/day) and methotrexate (0.3–0.5 mg/kg/week) also have a long history of use for cutaneous and extracutaneous sarcoidosis [56–59]. The results of a randomized controlled trial demonstrated that methotrexate is efficacious as a corticosteroid-sparing agent for new-onset sarcoidosis [60].
More recently, minocycline and pentoxifylline were found to be beneficial in open-label clinical trials of adults with cutaneous [61] and pulmonary [62] sarcoidosis, respectively. There are reports of improvement of cutaneous sarcoidosis with topical tacrolimus, phototherapy (ultraviolet A1 or psoralen plus ultraviolet A), isotretinoin and allopurinol [55–57,63–65]. Pulsed dye and carbon dioxide laser therapy may be helpful for recalcitrant skin lesions, but induction of ulceration or the development of new lesions has been observed [55,66]. Additional immunomodulatory agents that have been successfully used for systemic or severe recalcitrant cutaneous sarcoidosis include azathioprine, mycophenolate mofetil, anti-TNF agents (e.g. infliximab, adalimumab and etanercept), thalidomide and leflunomide [55–57,67,68]. Paradoxically, the development of sarcoid-like granulomas has also been described as an adverse effect of anti-TNF agents. Studies of cyclosporine therapy for sarcoidosis have overall had disappointing results, and exacerbation of skin disease has been noted [55,57,69].
References
1 Shetty AK. Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheum 2008;6:16–26.
2 Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med 2007;357:2153–65.
3 Rosé CD, Arostegui JI, Martin TM et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheum 2009;60:1797–803.
4 Hoffmann AL, Milman N, Byg KE. Childhood sarcoidosis in Denmark 1979–1994: incidence, clinical features and laboratory results at presentation in 48 children. Acta Paedriatr 2004;93:30–6.
5 Byg KE, Milman N, Hansen S. Sarcoidosis in Denmark 1980–1994: a registry-based incidence study comprising 5536 patients. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:46–52.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


