Normal hair loss/growth in childhood
Hair loss
Hypertrichosis
Hair loss in childhood is usually fraught with overwhelming concern by parents that the condition will be permanent and/or leave psychological scars on the affected child. Conversely, physicians are more likely to focus on the potential relatedness of the hair loss to an underlying medical problem. The concerns of both are valid. There is great value in diagnosing a given case of childhood alopecia, as herein may be the necessary clue to an otherwise unfathomable multisystem illness or an explanation for an unexplained developmental delay. The treatment of hair loss presenting in childhood does include disorders for which no good therapy yet exists, but there are many conditions in which specific treatment can either reverse the hair loss or make the hair more manageable and, hence, more cosmetically acceptable.
Hypertrichosis specifically refers to hair density or length beyond the accepted limits of normal for a particular age, race and sex and does not imply, as does the term hirsutism, a particular distribution of hair or a hormonal aetiology. Hypertrichosis may be generalized or localized, and may consist of lanugo, vellus or terminal hair. The presence of hypertrichosis in a child may signify an underlying physical abnormality, an associated metabolic disorder, a genetic multifocal syndrome or merely a cosmetic problem.
This chapter presents an effective approach to the diagnosis of the various types of alopecia and hypertrichosis presenting in childhood. Aetiologies of hair loss or hypertrichosis presented in detail in other chapters will be mentioned only briefly and the reader is referred to these other sources of information.
Normal Hair Loss/Growth in Childhood
To fully understand hair loss or excess hair growth in childhood, a basic working knowledge of normal hair growth is necessary, including the embryology and cycling of hair. Hair development begins in utero at 9–12 weeks, with the follicular units composed of epidermally derived follicles and mesodermally derived papillae [1] (Fig. 148.1). By 18–20 weeks of gestation, fine lanugo hair (unpigmented, unmedullated fine hair, which may grow to several centimetres in length [2,3]) covers the scalp and proceeds to appear elsewhere in a cephalocaudal direction, eventually covering the entire fetus. This constitutes the first anagen (growth) wave, which is followed by telogen (resting phase) and, eventually, the actual shedding of the hair at the seventh or eighth month [2,4]. The lanugo hair is replaced by vellus hair on the body and vellus or terminal hair on the scalp. The transition wave from anagen to telogen in the occipital area is delayed, however, and the expected telogen shedding in the occiput occurs at 2–3 months postpartum [2,4], accounting for the occipital alopecia normally seen in infants of this age (Fig. 148.2). Lanugo hair may also be seen on the limbs and shoulders of full-term, normally developed newborns, but this should be shed by 1–2 months of age.
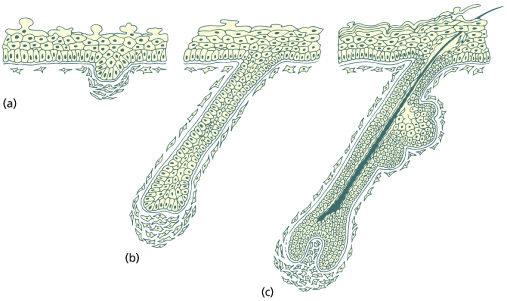
Fig. 148.1 Embryology of the hair follicle. (a) Follicular germ illustrating the condensing mesenchyme proximal to the epidermally derived follicle cells. (b) Follicle peg stage illustrating the organization of keratinocytes in the follicle and the mesenchyme of the follicle sheath and presumptive dermal papillae. (c) Bulbous hair peg stage illustrating the regions of the differentiated follicle. The upper bulge on the right represents the sebaceous gland and duct. The ‘bulge’ area where the arrector pili muscle will insert is below this.
For the remainder of the first year of life, scalp hair growth is synchronous, taking on the adult mosaic pattern only towards the end of the first year [2]. The number of follicles does not change after birth but, rather, the follicular density decreases as the skin expands to cover an increasing surface area [4,5]. There is a gradual transition from vellus (unmedullated, lightly pigmented hair, final length less than 2 cm [5]) to terminal (usually pigmented, usually medullated, generally thicker shafts with longer anagen phase and thus longer ultimate length) hair over the scalp during the first year or two. Hair colour tends to darken with age [6].
All human scalp hairs regularly cycle through various stages of growth. In anagen, or the active growth phase, the follicular bulb embraces the dermal papillae in the dermis or subcutaneous tissue (Fig. 148.3). The division and maturation of the matrix (those cells in the centre of the hair bulb contiguous to the dermal papillae) produce columns of cells that stream superficially into the central portion of the bulb and then enter the straight linear portion of the follicle [7]. The cellular keratin filaments (protein) organize into larger aggregates, which become progressively more compact as the shaft moves upwards and away from the bulb [8]. For much of the length of the follicle, the hair shaft is attached to layers of root sheaths, which serve both to anchor and to mould the newly formed hair. Anagen lasts for predetermined periods of time based on the area of the body the hair resides in; the normal time in anagen for scalp hair is longer and more variable than other body areas [9].
Fig. 148.3 (a–d) Normal cycling of hair. Follicular structures above the dashed line form the permanent part of the follicle. Epidermally derived cells below the bulge (B) degenerate during catagen and telogen. Note that, during catagen, the dermal papillae (DP) lags behind the ascending terminal bulb and that cells in the bulge region are poised for downward proliferation in early anagen (anagen II). When the germinative epithelium is once again in close approximation to the dermal papillae, the anagen growth cycle begins again. The preceding hair is lost as the new hair begins its growth phase. APM, arrector pili muscle; C, cortex; E, epidermis; IRS, inner root sheath; M, matrix; Md, medulla; ORS, outer root sheath; S, sebaceous gland.
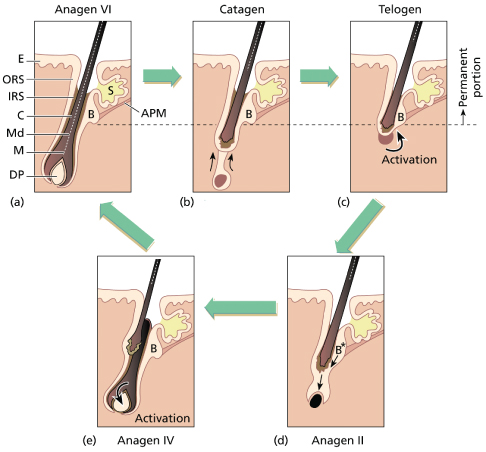
When a particular hair has completed its sojourn in anagen, it begins the process of transition to a resting (telogen) hair. The follicular bulb moves up in the dermis, with the dermal papillae no longer intimately associated but lagging behind (Fig. 148.3). The transition phase between the end of anagen and the beginning of telogen is referred to as catagen and lasts 2–4 weeks. With the loosening of the attachment of the root sheaths to the hair shaft, the telogen hair is now subject to being dislodged by simple pressure or traction. Once telogen is completed, and the time in telogen varies with body site, the cycle is repeated. Telogen in the scalp is normally 3–4 months. Cells in the outer root sheath of the follicle at the base of the arrector pili insertion, the bulge region, are slow-cycling stem cells that are necessary for the recapitulation of anagen; this involves a downward growth of the follicle and regeneration of the matrix and lower follicle root sheaths [9,10]. The spontaneous loss of a telogen hair, termed exogen [11], generally signals the presence in the follicular canal of a new growing anagen hair.
References
1 Pinkus H. Embryology of hair. In: Montagna W, Ellis RA, eds. The Biology of Hair Growth. New York: Academic Press, 1958:1–32.
2 Barth JH. Normal hair growth in children. Pediatr Dermatol 1987;4:173–84.
3 Danforth CH. Studies on hair. Arch Dermatol Syph 1925;11:804–21.
4 Barman JM, Pecoraro V, Astore I et al. The first stage in the natural history of the human scalp hair cycle. J Invest Dermatol 1967;48:138–42.
5 Giacometti L. The anatomy of the human scalp. In: Montagna W, ed. Advances in Biology of Skin, Vol. 6. Oxford: Pergamon Press, 1965:97–120.
6 Price ML, Griffiths WAD. Normal body hair: a review. Clin Exp Dermatol 1985;10:87–97.
7 Abel E. Embryology and anatomy of the hair follicle. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. New York: McGraw-Hill, 1994:1–19.
8 Bertolino A, O’Guin WM. Differentiation of the hair shaft. In: Olsen EA, ed. Disorders of Hair Growth: Diagnosis and Treatment. New York: McGraw-Hill, 1994:22–5.
9 Lyle S, Cristofidou-Solomidou M, Liu Y et al. The C8/144B monoclonal antibody recognizes cytokeratin 15 and defines the location of human hair follicle stem cells. J Cell Sci 1998;111:3179–88.
10 Oshima H, Rochat A, Kedzia C et al. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell 2001;104:233–45.
11 Milner Y, Sudnik J, Filippi M et al. Exogen, the shedding phase of the hair cycle: characterization of a mouse model. J Invest Dermatol 2002;119:639–44.
Hair Loss
Evaluation of the Child with Hair Loss
History.
The evaluation of a child with scalp hair loss should always include a history, physical examination and microscopic examination of the hair bulb and/or shaft. The history should differentiate hair never coming in fully from hair that once covered the scalp but was later lost or shed. However, it is entirely within the range of normal for either the so-called second pelage (i.e. the second wave of anagen scalp hair) or the transition from vellus to terminal hair to be delayed up to 1 year of age, making it falsely appear that the affected child has congenital alopecia.
Diffuse scalp hair loss that has a hereditary basis usually manifests itself by the first or second year of life, but in some genetically based disorders the associated hair loss becomes obvious only later (e.g. dyskeratosis congenita [1], Jorgensen syndrome [2], Beare pili torti [3], androgenetic alopecia). Obviously, family history is key in determining the exact mode of inheritance of a suspected genetic disorder, but family members may have only some, but not all, of the features associated with a particular syndrome, and alopecia may not be one of them. Therefore, when suspecting a familial syndrome, or probing for one that has alopecia as one feature, multiorgan signs and symptoms should be enquired about. As the group of disorders collectively called ectodermal dysplasias commonly involve hair loss (and effects on the teeth, nails and sweating), this should be looked for in particular. Dental radiographs may be necessary to exclude tooth involvement in the very young, and formal sweat testing may be necessary to document decreased sweating. Currently, there is no standardized test protocol for the evaluation of sweating in ectodermal dysplasia syndromes, but recommended techniques are those that assess both sweat gland number and function post sweat induction [4].
Physical Examination.
A physical examination should be performed in all children with hair loss of uncertain aetiology. The possibility of a syndrome must be entertained and multisystem abnormalities sought for and catalogued; those of ectodermally derived organs (epidermis-derived teeth, ears, eyes, central nervous system, mammary glands), bone, cleft lip and/or cleft palate are frequently associated with scalp hair loss. Particular attention should be paid to the child’s facial features and whether a distinctive facies is present.
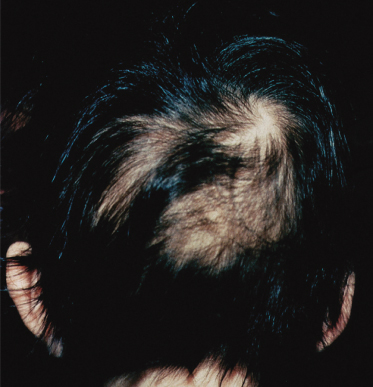
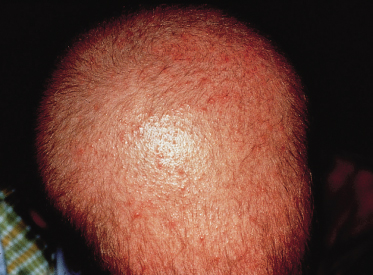
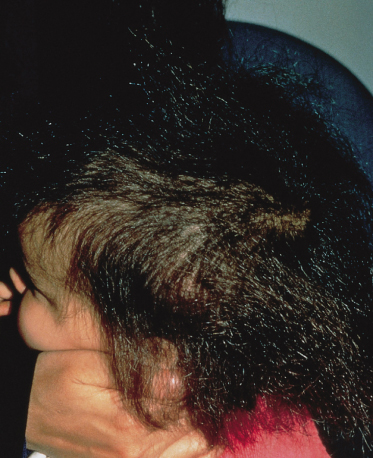
The scalp examination should first conclude whether the hair loss is diffuse (or global) or focal (Fig. 148.4). A diffuse loss could be secondary to an inherited abnormality in follicular or hair shaft development or an acquired problem such as alopecia areata, anagen effluvium or telogen effluvium. Focal alopecia is less likely to be inherited and much more likely to be acquired (Fig. 148.5a).
Fig. 148.4 Diffuse (global) hair loss in a child with Rosselli–Gulienetti syndrome (palate–popliteal pterygia syndrome) with subgroup type 1, 2, 3 and 4 ectodermal dysplasia.
Fig. 148.5 (a) Focal, non-scarring hair loss of alopecia areata. (b) Scarring (permanent) hair loss in a 10-year-old child, with dyspigmentation and follicular papules.
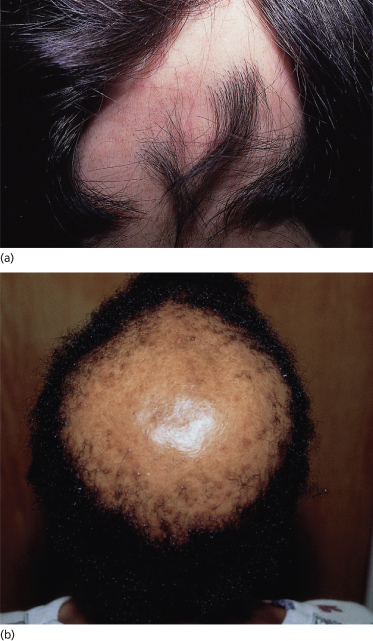
The scalp in the areas of alopecia should be evaluated for the preservation of follicular openings (implying a non-scarring potentially reversible process; Fig. 148.5a) compared with smooth, poreless skin indicating attrition of follicular units and a potentially irreversible or scarring process (Fig. 148.5b). Scarring alopecia is rare in children in the absence of congenital focal scalp abnormalities, tumours or trauma.
Further differentiation can be made between hair growth abnormalities secondary to: (i) failure to initiate anagen; (ii) hair fragility leading to breakage; (iii) unruly hair; and (iv) premature curtailment or interruption of anagen (abnormality of hair cycle), leading to increased hair shedding. To determine if the hair growth rate is affected, a simple hair window can be performed. In this procedure, hair is clipped flush with the scalp in an arbitrarily determined target area (generally at the back of the scalp to prevent manipulation by the patient) and in a shape unlikely to occur naturally (such as a square or rectangle) and the hair length in this area observed a few weeks later. Even if there is an underlying hair shaft fragility problem that precludes the hair growing long, the hair should be able to attain a length of 0.5 cm in 2–3 weeks (1 cm/month).
To determine whether abnormal shedding is occurring, a simple hair pull is performed. Approximately 50–100 hairs are grasped at the base between the thumb and forefinger and gently pulled proximally to distally. This procedure is repeated in various sections of the scalp, six to eight times in total. The number and type of shed hairs are counted: there should normally be only telogen hairs unless the patient is a very young child or there is underlying pathology. One to two anagen hairs on a hair pull in a young child is common, and most of these hairs assume the microscopic appearance of ‘loose anagen’ hairs [5], which are devoid of the attached root sheaths characteristic of anagen hairs. Loose anagen hairs on a hair pull in postpubescent children should trigger consideration of underlying hair pathology.
Microscopic Examination.
If increased hair shedding is present, the shed hairs collected by a hair pull should be examined under the light microscope. One to two drops of cyanoacrylic are placed on a slide and the proximal hair shafts/bulbs are lined up in the glue and a coverslip placed over them: this decreases distortion and provides a permanent record of the hairs in question. The bulbs are then examined to determine if the hair loss is telogen or anagen. Telogen bulbs are unpigmented, rounded up and devoid of an attached root sheath (Fig. 148.6a). Normal anagen hairs are not readily obtained on a hair pull test but if one or two are, they generally have attached root sheaths (Fig. 148.6b). Loose anagen hairs, which can be seen in either normal young children or in patients with loose anagen syndrome (Fig. 148.6c) or other causes of anagen hair loss such as alopecia areata, are devoid of root sheaths and have a ruffled or floppy sock appearance of the attached cuticle. Telogen versus anagen shedding should trigger very different types of work-up.
Fig. 148.6 (a) Telogen bulb (light micrograph, ×40); (b) anagen bulb (light micrograph, ×40); (c) loose anagen syndrome (light micrograph, ×100).
Reproduced from Olsen EA. Clinical Tools for Assessing Hair Loss. In: Olsen EA, ed. Hair Disorders: Diagnosis and Treatment. New York, McGraw-Hill, 1994.

If there is no abnormal shedding, but instead the hair fractures with simple trauma (rubbing the hair between the fingers is one way of precipitating this in susceptible patients), or has an abnormal texture or dullness resulting in unruliness, a sample of affected hairs should be clipped and the distal portion examined under the microscope. Most hair shaft abnormalities can be diagnosed in this manner, although some will require further examination by scanning electron microscopy to confirm findings only hinted at under light microscopy (e.g. longitudinal grooving). Polariscopic examination is necessary in cases when the particular light microscopic findings of trichoschisis with or without trichorrhexis nodosa are seen, making the diagnosis of trichothiodystrophy a possibility. The aetiology of brittle hair can be further pursued by chemical analysis of the hair for sulphur content and/or quantification of individual amino acids.
Together, these simple tools (history, physical examination and microscopic examination of the hair) will help narrow the differential diagnosis of alopecia. Dermoscopy, if available, may also be used to help establish the diagnosis in some hair disorders. The various aetiologies of childhood alopecia are discussed below in further detail.
Types of Hair Loss
Abnormality in Initiation of Hair Growth
Diffuse Loss
We are only now beginning to understand the genetic abnormalities associated with hair disorders. In several conditions, near or complete universal atrichia may be present at birth, or develop within the first 1–2 years of life. Caution should be exercised to ensure that the hair abnormality is isolated, as other associations may be unveiled only with time. For example, patients with mutations in the hairless gene may present with scalp alopecia alone [6] but develop characteristic keratin-filled epithelial cysts 3–18 years after the alopecia [7,8]. This syndrome, (atrichia with papular lesions), is generally an autosomal recessive trait [9]. Total alopecia related to the hairless gene without other associated findings has been reported to be autosomal dominant, autosomal recessive or X-linked [6,10]. Patients with autosomal recessive hereditary vitamin D-dependent rickets (VDDRII) also present with total or near total hair loss within the first year of life but later develop rickets and cutaneous cysts [11]. The genetic abnormality is a mutation in the vitamin D receptor. Universal alopecia may also occur with mental retardation and either talipes [12] or seizures [13–15]. Patients with the X-linked (Xq27.3–qter region) recessive condition Mendes da Costa–van der Valk genodermatosis present with universal alopecia at birth, or within the first few months of life, accompanied by reticular brown-red pigmentation on the face and extremities [16,17]. During the first few years of life, these patients develop recurrent non-traumatic intraepidermal blisters and may have associated acrocyanosis, microcephaly with mental retardation, dwarfism, short conic fingers and nail dystrophy [16,18].
Conditions in which follicular hyperkeratosis may be associated with total alopecia in infancy are presented in Table 148.1.
Table 148.1 Follicular hyperkeratosis with scalp alopecia
Modified from Olsen 2003 [19].
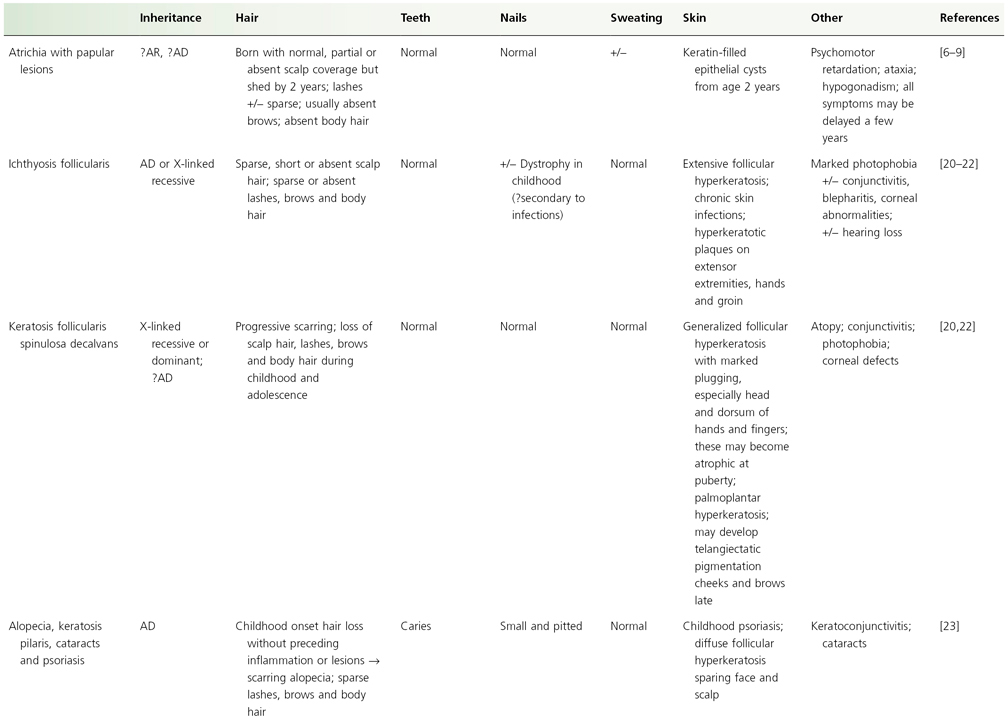
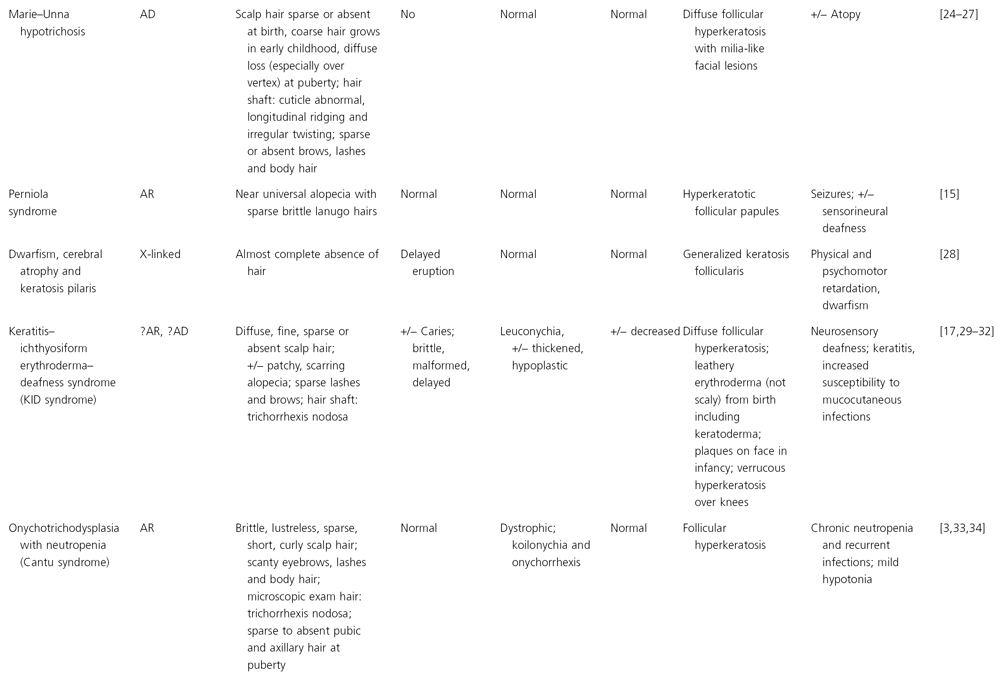
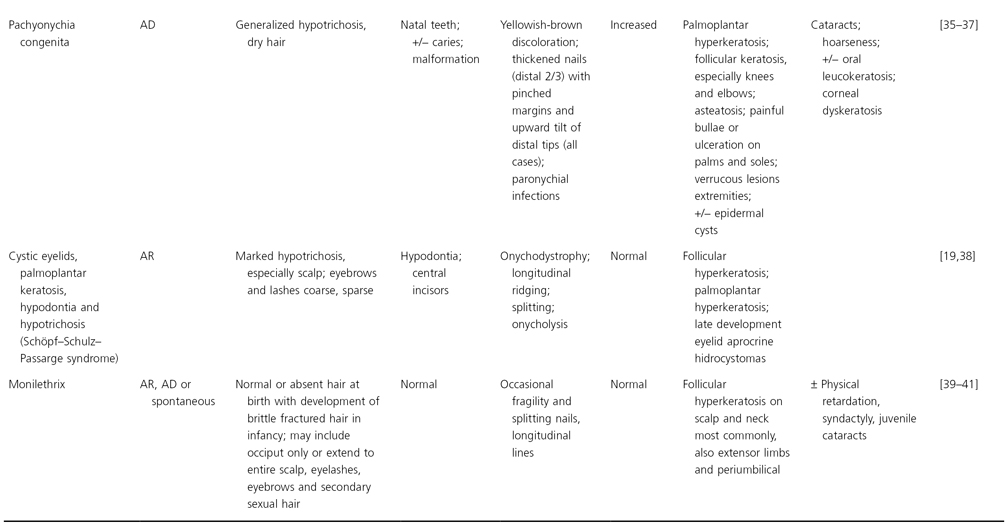
AD, autosomal dominant; AR, autosomal recessive.
A few of the ectodermal dysplasias that present with universal or near total alopecia in infancy include the following:
- Those associated with hair, nail and sweating abnormalities, e.g. odonto-onychodysplasia with alopecia [2,42], Hayden syndrome [2], alopecia–onychodysplasia–hypohidrosis syndrome [2,43], ectodermal alopecia with severe mental retardation [2], dermotrichic syndrome [2] and ectodermal dysplasia/skin fragility syndrome (McGrath syndrome), caused by a mutation in the plakophilin-1 gene [17].
- Those associated with hair, teeth and sweating abnormalities under the hypohidrotic ectodermal dysplasia phenotype and related to genetic anomalies in the signaling cascade that leads to nuclear factor kB (NF-kB) activation. These include autosomal recessive or autosomal dominant mutations in the EDAR (ectodysplasin receptor) gene or EDAR-associated death domain gene EDARADD, respectively, or X-linked defects in the ectodysplasin A (EDA-1) gene, the receptor for EDA-2 and the NF-kB essential modulator (NEMO), the latter also associated with immune deficiency [44].
- Those associated with hair and teeth abnormalities, e.g. alopecia, unusual facies and preaxial polydactyly (Wilson syndrome) [45].
- Those associated with hair and nail abnormalities, e.g. tricho-onychodysplasia with xeroderma [3,46], skeletal anomalies with ectodermal dysplasia and growth and mental retardation [2,47], cataracts–alopecia–sclerodactyly [48] and pure hypotrichosis and nail dysplasia [49].
Alopecia areata is the only potentially completely reversible universal alopecia that may present in infancy. This is rare in the first year of life.
There is a very long list of conditions that present with hypotrichosis, but not complete alopecia, in infancy. The hypotrichosis may be secondary to follicular hypoplasia or to faulty hair shaft production and breakage. Hypotrichosis simplex of the scalp, related to a mutation in the gene that encodes corneodesmosin, begins in the mid first decade and is associated with almost complete hair loss by the third decade [17]. Individuals with autosomal recessive localized hypotrichosis (scalp hair, largely sparing secondary sexual hair) may have sparse hair at birth that regrows poorly or not al all: this may be related to either a mutation in the LIPH (607365) gene on chromosome 3q27, desmoglein 4 on chromosome 18q12 or P2RY5 on chromosome 13q14.12-q14.2 [17,50–52].
Many of the ectodermal dysplasias are associated with hypotrichosis but, unfortunately, most of the hair shaft abnormalities have not been well characterized; the abnormal hair is generally described clinically only as ‘brittle’, ‘sparse’ or ‘lustreless’. (The ectodermal dysplasias are discussed in detail in Chapter 127.) Other non-ectodermal dysplasia syndromes present in infancy with sparse, lustreless hair as one part of multiorgan abnormalities (e.g. cartilage–hair hypoplasia (mutation in the RMRP gene) [17,53], hypomelia–hypotrichosis–facial haemangioma [54] and regional choroidal atrophy and alopecia [55]). Most of the over 10 subtypes of orofaciodigital syndrome are autosomal recessive but type I is an X-linked dominant abnormality of the CXORF5 gene. The hair in the latter is either dry and wiry or demonstrates diffuse or a mosaic pattern of alopecia [17,56]. The diagnosis of the primary condition in these cases is rarely suggested by the hair abnormality, probably secondary to the dearth of available information on the hair.
There are, however, a number of conditions that can be diagnosed by microscopic evaluation of the hair shaft. Depending on the type of hair shaft abnormality, they generally present as fragile or unruly hair. These will be presented here according to their microscopic description.
Hair Shaft Abnormalities Presenting with Hair Breakage
Trichorrhexis Nodosa
The most common defect of the hair shaft leading to hair breakage is trichorrhexis nodosa [57]. The primary abnormality is a focal loss of the cuticle, which leads to exposed and eventually frayed cortical fibres [58,59]. This appears initially microscopically as a nodal swelling and is followed by fracturing and splaying of the exposed fibres in a fan-like array (Fig. 148.7). Trichorrhexis nodosa can occur in normal hair that has been abused by excessive repetitive exposure to chemicals or physical trauma but more commonly occurs in inherently weak hairs after trivial trauma (e.g. brushing, combing).

Although trichorrhexis nodosa can present at birth as an isolated problem [60] or with teeth and/or nail abnormalities [61], its presence in an infant or young child should trigger a search for an underlying metabolic problem. One association is with argininosuccinic aciduria, an autosomal recessive disorder of the urea cycle caused by an abnormality in gene 7cen-q11.2 [17] in which the absence of the enzyme argininosuccinase – which normally splits argininosuccinic acid into arginine and fumaric acid – leads to acidosis, hyperammonaemia, low serum arginine, increased serum and urine citrulline and argininosuccinic acid [62,63]. In these children, seizures and hepatomegaly may begin in infancy while symptoms of psychomotor retardation, ataxia and dull brittle hair (with microscopic trichorrhexis nodosa) may first manifest after the age of 2 years [64,65].
Citrullinaemia, in which there is an abnormality of the enzyme argininosuccinic acid synthetase [66], may also present with increased serum citrulline and low arginine. Children with citrullinaemia may present with a scaly skin eruption and hair fragility, with trichorrhexis nodosa and pili torti on microscopic examination of the hair [67–69].
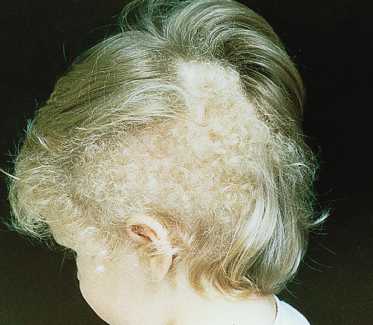
Patients with Menkes syndrome, or trichopoliodystrophy, an X-linked disorder of copper transport, also have trichorrhexis nodosa and pili torti on microscopic examination of the hair [70,71]. The defective gene, MKN or ATP7A, encodes a copper-translocating membrane protein adenosine triphosphatase (ATPase) that disturbs intracellular copper homeostasis and the function of copper-requiring enzymes [72,73]. Systemic copper deficiency occurs from trapping of copper in some tissues, particularly the intestine, kidney, fibroblasts and red blood cells, leading to failure of copper delivery to other tissues [74–77]. In affected children, the hair is normal at birth but is replaced in early infancy by sparse, brittle, depigmented hair that feels like steel wool, hence the colloquial term of ‘steely hair syndrome’ [77–79] (Fig. 148.8). The skin is characteristically pale and lax, the face expressionless and the child drowsy and/or listless. There may be associated hypothermia, mental retardation and degeneration of cerebral, cerebellar, bone and connective tissue. A low serum ceruloplasmin is diagnostic of Menkes syndrome. Treatment with copper is usually ineffective and most affected children die by the age of 3 years [80]. However, recent reports of immediate postpartum treatment with copper–histidine show a prevention or diminution in the severe neurodegeneration typical of the disease [72].
Trichoschisis
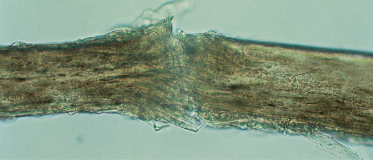
Trichoschisis is a clear transverse fracture through the entire hair shaft (Fig. 148.9). Under the light microscope, the affected hairs often appear flat and may be folded over as well [81]. Trichorrhexis nodosa may also be present. Under scanning electron microscopy, the areas of fracture are associated with localized absence of the cuticle [81]. Trichoschisis, although not absolutely pathognomonic, is nonetheless seen with regularity only in the condition termed ‘trichothiodystrophy’.
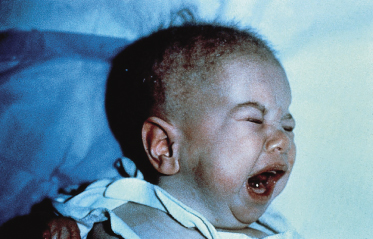
Trichothiodystrophy is an autosomal recessive disorder characterized by sulphur-deficient brittle hair which may occur alone or in conjunction with other neuroectodermal abnormalities [82]. The hair abnormality identifies a group of genetic disorders in which acronyms or eponyms identify particular constellations of extratrichological findings (Table 148.2). Clinically, patients with trichothiodystrophy have, since early infancy, short brittle hair on the scalp, eyelashes or eyebrows (Fig. 148.10). The cystine content of the hair is about one-half of normal, primarily due to a major reduction and altered composition of the high sulphur matrix proteins [100–103]. Polariscopic examination of affected hairs characteristically shows alternating dark and light bands (Fig. 148.11), presumably secondary to the alternating sulphur content [104]. Sulphur and/or amino acid analysis of the hair is diagnostic.
Table 148.2 Syndromes associated with trichothiodystrophy
Modified from Whiting 2003 [57].
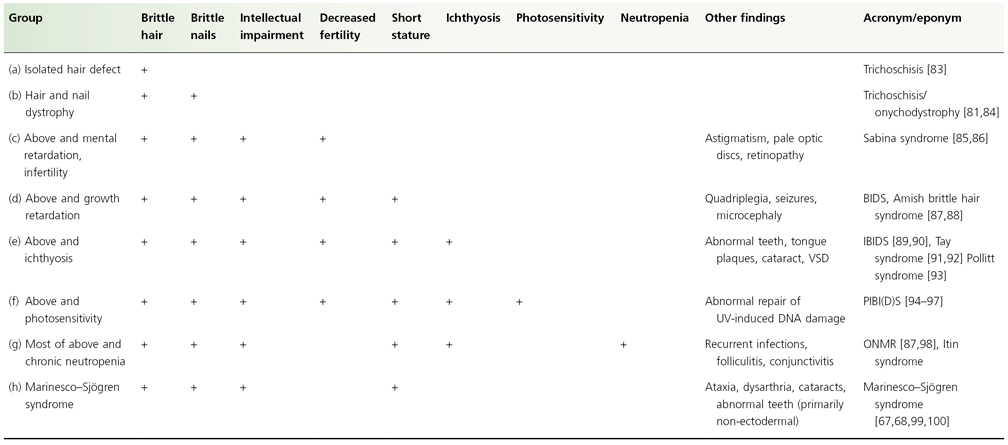
BIDS, brittle hair, impaired intelligence, decreased fertility and short shature; IBIDS, ichthyosis, brittle hair, impaired intelligence, decreased fertility and short stature; ONMR, onychotrichodysplasia, neutropenia, mental retardation; PIBI(D)S, photosensitivity, ichthyosis, brittle hair, impaired intelligence, (decreased fertility) and short stature; UV, ultraviolet; VSD, ventricular septal defect.
Fig. 148.11 Trichothiodystrophy (polariscopic micrograph, ×40). Note the alternating light and dark bands.
Courtesy of Dr David A. Whiting.

Other abnormalities should be sought in those patients with trichothiodystrophy (Table 148.2), particularly the presence of photosensitivity. Patients with trichothiodystrophy, particularly the 50% with photosensitivity, may have a defect in excision repair of ultraviolet damage but without an increased risk of skin cancer [105]. It has recently been determined that the various clinical presentations and DNA repair characteristics of both photosensitive trichothiodystrophy and xeroderma pigmentosum can be correlated with mutations found in the ERCC2/XPD locus on chromosome 19, with trichothiodystrophy due primarily to mutations that affect the transcriptional role of ERCC2 and xeroderma pigmentosum due to mutations that primarily alter the repair role of ERCC2 [17,105]. Photosensitive trichothiodystrophy is also uncommonly associated with mutations in gene ERCC3/XPB on chromosome 2 and TTD-A on chromosome 6. Both the XPD and XPB genes encode the two helicase subunits of transcription/repair vector TFIIH. The TTD-A gene is associated with a mutation in the 10th subunit of TFIIH. The non-photosensitive trichothiodystrophy mutation has been mapped to variations in the C70RF11 gene map locus 7p14 [17].
Trichorrhexis Invaginata
Trichorrhexis invaginata (bamboo hair) clinically presents in infancy with short, brittle, often sparse hair [106] (Fig. 148.12). The primary defect appears to be abnormal keratinization of the hair shaft in the keratogenous zone allowing intussusception of the fully keratinized and hard distal shaft into the incompletely keratinized and soft proximal portion of the shaft [107,108]. This leads to a proximal cup-like or socket-like expansion embracing a ‘ball’, the typical ‘ball and socket’ deformity (Fig. 148.13). Fracture of the shaft through this area is common, but there may also be disarticulation of the distal ‘ball’, leaving a golf-tee or tulip-shaped end to the abnormal hair [109] (Fig. 148.14). These changes may be seen with dermoscopy but should be confirmed microscopically. Pili torti and trichorrhexis nodosa may also be seen with trichorrhexis invaginata. Sulphur content is normal in trichorrhexis invaginata and no scanning electron microscopic studies are necessary to make this diagnosis. However, the abnormal hairs may be present only in some sections of the scalp, so many areas of the scalp (or even eyebrows) may need to be evaluated to make a definitive diagnosis.
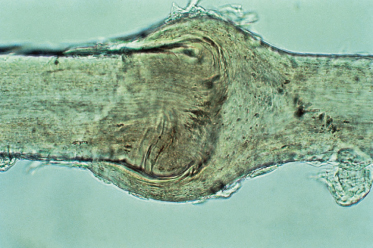
Fig. 148.14 Trichorrhexis invaginata, golf-tee fracture (light micrograph, ×200)
Reproduced from Whiting 2003 [57].

Trichorrhexis invaginata can rarely occur in traumatized, otherwise normal hair or with other congenital hair shaft abnormalities. Usually, however, the hair abnormality is associated with Netherton syndrome, an autosomal recessive inherited disorder that consists of the triad of ichthyosis, atopic diathesis and trichorrhexis invaginata [110–112]. The ichthyosis is most commonly ichthyosis linearis circumflexa, a polycyclic, ever-transforming scaly eruption with a double-edged scale on the leading edge [108,113] (Fig. 148.15]. However, some cases of trichorrhexis invaginata have instead been associated with lamellar ichthyosis or, less commonly, ichthyosis vulgaris or X-linked ichthyosis [112,114]. The atopic diathesis usually includes persistent xerosis and may include erythroderma [111,115]. The diagnosis of Netherton syndrome should always be entertained in ‘red scaly babies’ who have sparse hair. Recurrent infections, short stature and mental retardation have been reported rarely in Netherton syndrome [116]. The Netherton gene, a mutation in the gene for the serine protease inhibitor, SPINK5, has recently been localized to chromosome 5q32 [117]. The defect in the skin barrier seen in Netherton syndrome may be secondary to proteolysis, whereas the infections and atopic diathesis may be related to SPINK5-related effects on T-lymphocyte maturation and response [118].
There is no specific treatment for trichorrhexis invaginata. Retinoids and photochemotherapy have been reported to be of some value and the condition may spontaneously improve with age [119–121].
Pili Torti
Patients with pili torti typically present with short, brittle hair. Microscopically, the hair is flattened and twisted on its own axis, anywhere from 90° to 360° [122]. Twisted hairs on the scalp may normally be seen sporadically in Caucasians and are the norm in people of African descent and in the pubic/axillary hair of both races. For pili torti to be diagnosed, there must be multiple twists at irregular intervals on a given hair (Fig. 148.16). The affected hairs generally fracture through the twists.
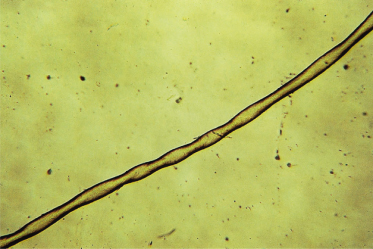
Pili torti, like trichorrhexis nodosa, can occur in the presence of other hair shaft abnormalities as either an inherited or an acquired finding and is also present in many different syndromes. It has been reported to occur in association with monilethrix [123], pseudomonilethrix [124], woolly hair [125], longitudinal grooving [126], trichorrhexis nodosa [59] and trichorrhexis invaginata [138]. In the classic Ronchese type of pili torti, the inheritance is usually autosomal dominant, but autosomal recessive and sporadic inheritance have also been reported [122,128–130]. The hair abnormality usually presents in infancy [127]; however, as with many inherited hair abnormalities, the first and second pelages may be normal with the pili torti not developing until the second year. Pili torti may be an isolated finding or part of an ectodermal dysplasia complex of findings. Sensorineural deafness has been described in a number of cases, and early auditory testing should be carried out in all children with pili torti [131,132]. Pili torti may also be present in many other syndromes, which are summarized in Box 148.1. The hair abnormality in these conditions may persist indefinitely or improve at puberty.
Box 148.1 Infantile Hair Loss Associated with Pili Torti
- Ectodermal dysplasia:
- Rapp–Hodgkins syndrome
- Solamon syndrome
- Arthrogryposis and ectodermal dysplasia
- Ectodermal dysplasia with syndactyly
- Tricho-odonto-onychodysplasia with pili torti
- Pili torti and enamel hypoplasia (Ronchese type)
- Pili torti and onychodysplasia (Beare type)
- Ankyloblepharon–ectodermal defects with cleft lip and palate syndrome
- Rapp–Hodgkins syndrome
- Björnstad dysplasia
- Salti and Salem syndrome
- Crandall syndrome
- Menkes kinky hair syndrome
- Tay syndrome and other cases of trichothiodystrophy
- Chondrodysplasia punctata
- Bazex syndrome
- Hypotrichosis with juvenile macular dystrophy [133]
- Citrullinaemia
Modified from Olsen, 2003 [19].
Pili torti may also present as a focal area of abnormal hair. This is usually secondary to trauma or to an underlying scarring condition of the scalp.
Monilethrix
Macroscopically, the hairs of monilethrix appear beaded, and with dermoscopy look like a ‘regularly bended ribbon’ [134]. Microscopically there are elliptical nodes occurring with regular periodicity every 0.7–1 mm [57] (Fig. 148.17). In between the nodes, the hair shaft is constricted, and it is at these points that the hairs usually fracture. Pili torti is often mistaken for monilethrix by the uninitiated because of the microscopic illusion of variation in diameter of the shaft due to twisting. On scanning electron microscopy of monilethrix hairs, there are structural abnormalities of both the cortex and cuticle in the zone of keratinization [135].

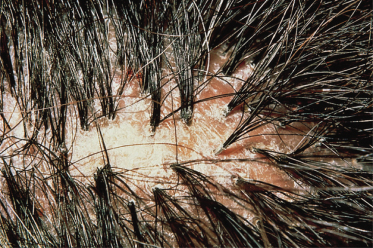
Most pedigrees show autosomal dominant inheritance with high penetrance [123,136] and in these cases the disorder has been found to be closely linked to the type II keratin gene cluster on chromosome 12q13, implicating a mutation in the structure or regulation of a trichocyte keratin gene in the pathogenesis of this disorder [137]. Recessively inherited cases have been reported and are related to defects in the desmoglein 4 (DSG4) gene, the same locus as autosomal recessive hypotrichosis [39]. The condition may result in delayed onset of hair loss with presentation at any time from infancy to the teens. Expression is variable with a spectrum of localized to global alopecia [137]. The clinical picture of monilethrix, however, can be very distinctive secondary to the appearance of extremely short brittle hairs emerging through keratotic follicular papules (Fig. 148.18). The occiput and nape of the neck are especially affected.
The hair defect may occur alone or in association with keratosis pilaris, physical retardation, syndactyly, cataracts and nail/teeth abnormalities [138]. Improvement in hair brittleness may occur during the summer and with age [34]. Etretinate/acitretin and topical minoxidil may potentially be useful therapies [41,139–141]. Protection against trauma such as excessive brushing, styling and braiding is key to limiting hair breakage.
Pseudomonilethrix
Pseudomonilethrix is microscopic irregular beading along the hair shaft as opposed to the regular beading seen in monilethrix [124] (Fig. 148.19). Although it has been reported in patients with fragile hair [124,142], the appearance of pseudomonilethrix can be produced in normal hairs by compressing two hairs together between two glass slides [143,144]. It is likely that the nodes in pseudomonilethrix are artefactual [57].
Fig. 148.19 Pseudomonilethrix (light micrographs, ×200). (a) Pseudomonilethrix induced by pressure on overlapped normal hairs. (b) Image rotated 90° to demonstrate indentation.
Reproduced with permission from Björnstad, 1965 [131].

Hair Shaft Abnormalities Associated with Unruly Hair
Uncombable Hair Syndrome
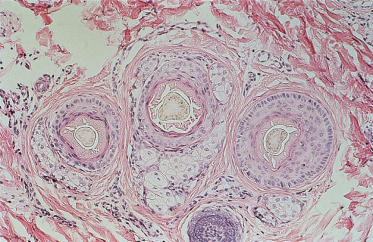
Children with uncombable hair syndrome present in infancy up to puberty with slow growing, silvery-blond ‘spun-glass’ hair that is disorderly and unmanageable [145–148] (Fig. 148.20). Under light microscopy, the hairs may appear normal or may have some midline darkening suggestive of the typical longitudinal grooves so clearly seen on scanning electron microscopy [149–151]. Longitudinal grooving in itself is a relatively common hair shaft abnormality, being seen in normal hair and in many cases of ectodermal dysplasia along with other hair shaft abnormalities [57]. On scanning electron microscopy of hairs in the uncombable hair syndrome, the longitudinal grooving is generally seen in conjunction with a cross-sectional triangular shape, the basis for the term pili trianguli et canaliculi [148] (Figs 148.21 and 148.22). One potential explanation for the hair shaft abnormality is premature keratinization of the inner root sheath: normally, the inner root sheath forms a rigid casing that influences the resultant shape of the hair shaft (normally round or oval) [146]. By itself, pili trianguli et canaliculi does not lead to hair fragility.
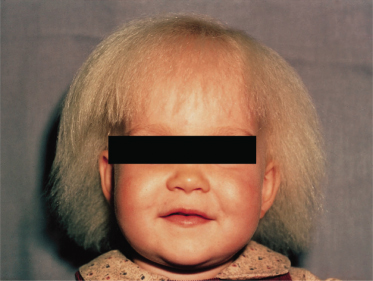
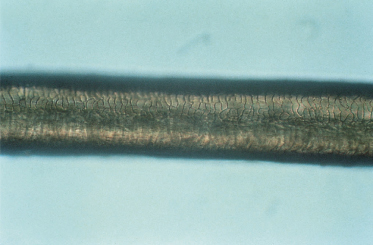
Fig. 148.22 Cross-section of hairs on a scalp biopsy of a child with uncombable hair syndrome. Note the triangular cross-section of an affected hair (horizontal section, haematoxylin and eosin stain).
Courtesy of Dr David A. Whiting.
The condition may be sporadic or exhibit autosomal dominant inheritance [149,152] and is generally without other associations or abnormalities. The condition of uncombable hair syndrome may improve with age. Supplemental biotin has been reported to be of use in one case [153] but generally does not affect the process. Conditioners are helpful.
Woolly Hair
Woolly hair is the presence of Negroid hair on the scalp of persons of non-Negroid descent. Microscopically, the hair is tightly coiled without generally going to the extremes of pili torti. However, pili torti and pili annulati (blond hair with both the clinical and microscopic findings of alternating bands of light and dark on the hair shaft) may be seen with this condition [125].
The hair in woolly hair is unruly only in the sense that it is difficult to manage, but probably not more so than the hair normally occurring in dark skinned persons of African descent. The aberrant hair growth begins at birth or infancy with excessively tight curls, making the hair appear bushy or frizzy. Hair length may be decreased secondary to brittleness, a common problem with Negroid hair in general. Woolly hair may go from curly to wavy as the child ages.
Woolly hair usually appears as a solitary problem inherited in an autosomal dominant fashion [125] but has been reported in conjunction with enamel hypoplasia [154], ocular defects [155,156], deafness and ichthyosis vulgaris [157], keratosis pilaris atrophicans [158] and Noonan syndrome [159]. Woolly hair, keratoderma and various cardiac abnormalities have been reported in Naxos syndrome (mutation in the plakoblobin gene, gene map locus 17q21, arrhythmogenic right ventricular cardiomyopathy) and two syndromes associated with a mutation in the gene encoding desmoplakin, gene map locus 6p24, i.e. Carvajal syndrome (dilated cardiomyopathy) and the Naxos-like syndrome (arrhythmogenic right ventricular dysplasia) [17,57,160]. Skin fragility and woolly hair without cardiac abnormalities have been reported with another mutation of the desmoplakin gene [17]. Woolly hair has been noted to occur in a sporadic recessive form and, in these cases, the scalp hair may be fine and white-blond and may lead to severe universal hypotrichosis in childhood: mutations have been associated with gene P2RY5 [17,125,161,162]. With excessively curly hair in a non-Negroid infant, one must also consider the following syndromes: trichodento-osseous syndrome (small widely spaced teeth, frontal bossing and dolichocephaly) [163] and CHAND (curly hair, ankyloblepheron and nail dysplasia) syndrome [164].
Marie–Unna Type of Hereditary Hypotrichosis
This autosomal dominant inherited condition has a distinctive type of hair loss that varies with the child’s age [23–26]. The hair is sparse or absent at birth with variable abnormal coarse scalp hair regrowth in childhood and potential scalp hair loss again at puberty (Fig. 148.23). There is associated general hypotrichosis of body hair. The coarse, wiry, twisted hair is very distinctive. Hair shaft examination shows irregular twisting and, on scanning electron microscopy, longitudinal ridging and peeling of the cuticle. Diffuse follicular hyperkeratosis with milia-like facial lesions may be present. A distinct gene has been noted close to the hairless gene in chromosome region 8p21 [165]. Recent work suggests a loss of function mutation of an inhibitory upstream ORF (U2HR) in the gene encoding the human hairless homologue [166].
Additional causes of wiry hair in childhood that can be lost after puberty include those conditions related to defects in the TP63 gene at 3q27. These all are characterized by hypohidrosis and cleft lip/palate and have been designated by the other consistent abnormalites present (Rapp–Hodgkin (none), EEC3 (ectrodactyly) and AEC (ankyloblepharon)) [17,167].
Acquired Localized Unruly Hair
Four non-inherited conditions may present as patches of scalp hair that differ from the normal texture/quality of hair for that individual. The most common is X-ray therapy related, with the hair that regrows after treatment (and epilation) being different in quality from that seen pretreatment. Localized woolly hair naevus, occurring only in non-Negroid persons, usually develops within the first 2 years of life (although this has been first reported in adolescence), with the affected hair being finer, lighter and more tightly curled than that of the rest of the scalp hair [168] (Fig. 148.24). Microscopically, the hairs may show trichorrhexis nodosa, longitudinal grooving, flattening and twisting [169–171]. Almost 50% of patients with woolly hair naevus have an underlying linear epidermal naevus or pigmented naevus, usually other than on the scalp [172,173].
Straight hair naevus, in which a localized portion of the normally curled or kinky hair is straight, has been noted only in Negroid persons. This may also have an association with an underlying epidermal naevus [174,175]. Acquired progressive kinking occurs after puberty, generally in males with androgenetic alopecia, and presents as gradual curling and darkening of the frontal, temporal, auricular and vertex hairs [176–179]. Microscopically, the hairs of acquired progressive kinking are short with kinks and twists and may show longitudinal grooving.
Localized Tufts of Hair
In pili multigemini, hairs from two to eight follicular bulbs, each with their own inner root sheath but surrounded by a common outer root sheath, emerge from one follicular canal [180]. In children, this condition may appear as an isolated scalp problem or may occur with classic pili torti [128] or in cleidocranial dysostosis [181]. Although compound follicles may appear similar to pili multigemini, in this condition two or three different hair shafts, each with their own inner root sheath and outer root sheath, eventually emerge from the same follicular opening. These two non-scarring entities must be differentiated from tufted folliculitis, in which scalp inflammation is prominent and leads to focal scarring with units of 10–15 hairs, each hair from its own follicle, emerging as tufts of hair from a single follicular canal [182,183] (Fig. 148.25).
Fig. 148.25 Tufted folliculitis.
Reproduced from Tong and Baden 1989 [183] with permission from Elsevier.
Abnormal Cycling
For the purposes of facilitating diagnosis, there are two main outcomes of premature disruption of anagen and, hence, there are two types of hair loss – anagen loss or telogen loss. Both should be suspected by the clinical presentation of abnormal shedding and confirmed by histological evaluation of the proximal hair shaft/bulb. The differential diagnosis and consequent evaluation and treatment vary greatly, however, between these two conditions.
Anagen Loss
Anagen Effluvium
Anagen loss is always abnormal and, with the exception of loose anagen syndrome and alopecia areata, scalp anagen hair loss generally implies a toxic exposure. The most common and easily recognizable cause of anagen loss (or effluvium) is X-ray therapy or chemotherapy. In both cases there may be a diminution of metabolic activity in the matrix, which results in weakening of the hair shaft, which breaks off a few millimetres from the scalp surface (Fig. 148.26
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


