The radiological findings of osteopoikilosis consist of multiple, well-circumscribed round or oval opacities, each 1–10 mm in diameter. They are usually found in the epiphyses and metaphyses of long bones and the pelvis, but are also frequent in the spongiosa of the phalanges, carpal and tarsal bones. The ribs, skull and spine are very rarely affected, which is helpful in excluding other osteocondensing conditions such as metastases, mastocytosis and tuberous sclerosis [22].
Osteopoikilosis is of no pathological significance and is usually an incidental finding, found in 12 of 211,000 radiographs in one series [38]. It can occur in the fetus, but usually takes many years to develop and may not be detectable before the late adolescent or adult period. Familial osteopoikilosis in the absence of skin changes has been described [39].
Buschke–Ollendorff syndrome usually remains a benign disorder throughout life, as exemplified by a woman who gave birth to eight affected children [27]. Rarely, muscle fibrosis and contractures may complicate the disorder [40] and several associations have been described [11,41], most of which are likely to be purely coincidental. One exception to this is the association with otosclerosis [5,23,42,43], possibly as a consequence of a generalized connective tissue disorder.
Differential Diagnosis and Treatment.
History, careful clinical examination and appropriate radiological investigations of the patient and whole family are essential to identify BOS. Indeed, the differential diagnosis of the cutaneous findings is quite large (Box 145.1). In the absence of bone changes in any member of the family, a biopsy will differentiate BOS from other connective tissue naevi.
Box 145.1 Differential Diagnosis of Cutaneous Elastoma in the Buschke–Ollendorff Syndrome
- Shagreen patch (tuberous sclerosis)
- Collagenoma
- Pseudo-xanthoma elasticum
- Lichen myxoedematosus
- Dermal nodules of Hunter syndrome
- Smooth muscle hamartoma
- Leiomyoma
- Neurofibroma
- Lipoma
The lesions of BOS remain asymptomatic and rarely cause cosmetic problems, so no treatment is necessary. Informing close relatives of the diagnosis is advisable to avoid misinterpretation of incidental radiographs and allow genetic counselling of this autosomal dominant syndrome.
Papular Elastorrhexis
Papular elastorrhexis is a rare variant of connective tissue naevus in which there is normal collagen and a decreased amount of elastic fibres. The disorder, appearing in adolescence, has been described in three single patients [44,45]. In these non-familial cases, the cutaneous findings were distinct from papular acne scars and not associated with extracutaneous abnormalities. Although some believe that most connective tissue naevi-like lesions, including papular elastorrhexis, in adults are papular acne scars [46], it seems that the distinctive histology of papular elastorrhexis clearly separates the condition from other entities [47]. Less than 15 cases were published in 2008 [48].
Schirren et al. described three members of one family presenting with non-follicular, distributed, white papules on the trunk and extremities [49]. The clinical appearance with absence of osteopoikilosis and the histological findings (decreased, fragmented elastic fibres and normal collagen) were compatible with papular elastorrhexis. However, on the basis of the genetic background, the authors believed that papular elastorrhexis was an abortive form of the Buschke–Ollendorff syndrome and suggested that connective tissue naevi with the most prominent alterations in the elastic tissue should be classified under the term elastic tissue naevi.
References
1 Buschke A, Ollendorff H. Ein Fall von Dermatofibrosis disseminata und Osteopathia condensans disseminata. Dermatol Wochenschr 1928;86:257–62.
2 Stieda A. (Ueber umschriebene Knochenverdichtungen im Bereich des Substantia spongiosa im Röntgenbilde. Bruns Beitr Klin Chir 1905;45:700–3.
3 Albers-Schoenberg HE. (Eine seltene, bisher nicht bekannte Strukturanomalie des Skelettes. Fortschr Roentgenstr 1915;23:174–5.
4 Berlin R, Hedensiö B, Lilja B et al. (Osteopoikilosis – a clinical and genetic study. Acta Med Scand 1967;181:305–14.
5 Cairns RJ. (Familial juvenile elastoma, osteopoikilosis (2 cases). Proc Roy Soc Med 1967;60:1267.
6 Grupper C, Cardinne A. Disseminated lenticular dermatofibrosis with osteopecila (father and son). Buschke–Ollendorff syndrome. Ann Dermatol Syphiligr Paris 1974;101:405–407.
7 Marshall J. Osteopoikilosis and connective tissue naevi: a syndrome of hereditary polyfibromatosis. S Afr Med J 1970;44:775–7.
8 Pastinszky J, Csato Z. On skin variations in osteopoikilia (Buschke–Ollendorff syndrome). Z Haut Geschlechtskr 1968;43:313–23.
9 Schorr WF, Opitz JM, Reyes CN. The connective tissue naevus – osteopoikilosis syndrome. Arch Dermatol 1972;81:249–52.
10 Smith AD, Waisman M. Connective tissue naevi: familial occurrence and association with osteopoikilosis. Arch Dermatol 1960;81:249–52.
11 Verbov J, Graham R. Buschke–Ollendorff syndrome – disseminated dermatofibrosis with osteopoikilosis. Clin Exp Dermatol 1986;11:17–26.
12 Morrison JG, Jones EW, MacDonald DM. Juvenile elastoma and osteopoikilosis (the Buschke–Ollendorff syndrome). Br J Dermatol 1977;97:417–22.
13 Weidman FD, Anderson NP, Ayres S. Juvenile elastoma. Arch Dermatol Syphil 1933;28:182–9.
14 Staricco RG, Mehregan AH. Naevus elasticus and naevus elasticus vascularis. Arch Dermatol 1961;84:943–7.
15 De Graciansky P, Leclerc R. Le ‘naevus elasticus’ en tumeurs disséminées. Ann Dermatol Syphil 1960;187:5–25.
16 Dammert K, Niemi KM. Naevus elasticus (elastoma juvenile Weidman) and naevus collagenicus lumbosacralis in Pringle’s disease. Dermatologica 1968;137:36–45.
17 Marguery MC, Samalens G, Pieraggi MT et al. Conjunctive nevus of the disseminated elastic type without osteopoikilosis or Weidman juvenile elastoma. Ann Dermatol Venereol 1991;118:465–8.
18 Huilgol SC, Griffiths WA, Black MM. Familial juvenile elastoma. Australas J Dermatol 1994;35:87–90.
19 Woodrow S, Pope F, Handfield-Jones S. The Buschke–Ollendorff syndrome presenting as familial elastic tissue naevi. Br J Dermatol 2001;144:890–3.
20 Ramme K, Kolde G, Stadler R. Dermatofibrosis lenticularis disseminata with osteopoikilosis. Buschke–Olldendorff syndrome. Hautarzt 11993;44:312–14.
21 Dahan S, Bonafe JL, Laroche M et al. Iconography of Buschke Ollendorff syndrome: X ray computed tomography and nuclear magnetic resonance of osteopoikilosis. Ann Dermatol Venereol 1989;116:225–30.
22 Roberts NM, Langtry JA, Branfoot AC et al. Case report: osteopoikilosis and the Buschke–Ollendorff syndrome. Br J Radiol 1993;66:468–70.
23 Schnur RE, Grace K, Herzberg A. Buschke–Ollendorff syndrome, otosclerosis, and congenital spinal stenosis. Pediatr Dermatol 1994;11:31–4.
24 Thieberg MD, Stone MS, Siegfried EC. What syndrome is this? Buschke–Ollendorff syndrome. Pediatr Dermatol 1993;10:85–7.
25 Trattner A, David M, Rothem A et al. Buschke–Ollendorff syndrome of the scalp: histologic and ultrastructural findings. J Am Acad Dermatol 1991;24:822–4.
26 Ghomrasseni S, Dridi M, Bonnefoix M et al. Morphometric analysis of elastic fibres from patients with: cutis laxa, anetoderma, pseudoxanthoma elasticum, and Buschke–Ollendorff and Williams–Beuren syndromes. J Eur Acad Dermatol Venereol 2001;15:305–11.
27 Al Attia H, Sherif A. Buschle–Ollendorff syndrome in a grande multipara: a case report and short review of the literature. Clin Rheumatol 1998;17:172–5.
28 Hellemans J, Preobradzhenska O, Willaet A et al. Loss-of-function mutations in LEMD3 result in ostoepoikilosis, Buschke–Ollendorff syndrome and melorheostosis. Nat Genet 2004;36:1213–18.
29 Mumm S, Wenkert D, Zhang X et al. Deactivating germline mutations in LEMD3 cause osteopoikilosis and Buschke–Ollendorff syndrome, but not sporadic melorheostosis. J Bone Min Res 2007;22:243–50.
30 Debeer P, Pykels E, Lammens J et al. Melorheostosis in a family with autosomal dominant osteopoikilosis: report of a third family. Am J Med Genet 2003;119:188–93.
31 Happle R. Segmentale Type-2-Manifestation autosomal dominanter Hautkrankheiten. Hautarzt 2001;52:283–7.
32 Ehrig T, Cockerell CJ. Bushke–Ollendorff syndrome: report of a case and interpretation of the clinical phenotype as a type 2 segmental manifestation of an autosomal dominant skin disease. J Am Acad Dermatol 2003;49:1163–6.
33 Gass JK, Hellemans J, Mortier G et al. Buschke–Ollendorff syndrome: a manifestation of a heterozygous nonsense mutation in the LEMD3 gene. J Am Acad Dermatol 2008;58:s103–4.
34 Giro MG, Duvic M, Smith LT et al. Buschke–Ollendorff syndrome associated with elevated elastin production by affected skin fibroblasts in culture. J Invest Dermatol 1992;99:129–37.
35 Hess W. Roentgenologische und pathologisch–anatomische Beobachtungen bei einem Fall von Osteopoikilie. Fortschr Geb Roentgenstr 1940;62:252–8.
36 Verbov J. Buschke–Ollendorff syndrome (disseminated dermatofibrosis with osteopoikilosis). Br J Dermatol 1977;96:87–90.
37 Atherton DJ, Wells RS. Juvenile elastoma and osteopoikilosis (the Buschke–Ollendorf syndrome). Clin Exp Dermatol 11982;7:109–13.
38 Jonasch E. 12 Faelle von Osteopoikilie. Fortrschr Roentgenstr 1955;82:344–53.
39 Sarralde A, Garcia Cruz D, Nazara Z et al. Osteopoikilosis: report of a familial case. Genet Couns 1994;5:373–5.
40 Walpole IR, Manners PJ. Clinical considerations in Buschke–Ollendorff syndrome. Clin Genet 1990;37:59–63.
41 Reid EM, Baker BL, Stees MA et al. Buschke–Ollendorff syndrome: a 32-month-old boy with elastomas and craniosynostosis. Pediatr Dermatol 2008;25:349–51.
42 Strosberg JM, Adler RG. Otosclerosis associated with osteopoikilosis. JAMA 1981;246:2030–1.
43 Piette Brion B, Lowy Motulsky M, Ledoux Corbusier M et al. Dermatofibromas, elastomas and deafness: a new case of the Buschke–Ollendorff syndrome. Dermatologica 1984;168:255–8.
44 Bordas X, Ferrandis C, Ribera M et al. (Papular elastorrhexis: a variety of nevus anelasticus? Arch Dermatol 1987;123:433–4.
45 Sears J, Seabury Stone M, Argenyi Z. Papular elastorrhexis: a variant of connective tissue nevus. J Am Acad Dermatol 1988;19:409–14.
46 Wilson B, Dent C, Cooper P. Papular acne scars. A common cutaneous finding. Arch Dermatol 1990;126:797–800.
47 Buechner S, Itin P. Papular elastorrhexis. report of five cases. Dermatology 2002;205:198–200.
48 Pajot C, Le Clec’h C, Hoareau F et al. Elastorrhexie papuleuse: deux cas. Ann Dermatol Venereol 2008;135:757–61.
49 Schirren H, Schirren C, Stolz W et al. Papular elastorrhexis: a variant of dermatofibrosis lenticularis disseminata (Buschke–Ollendorff syndrome)? Dermatology 1994;189:368–72.
Marfan Syndrome
Definition.
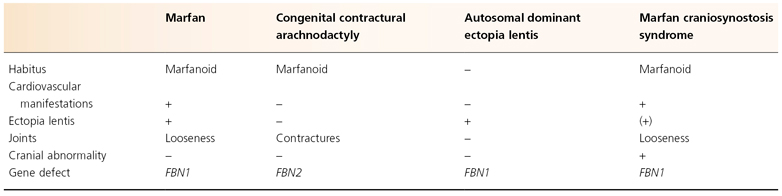
Marfan syndrome (MFS) is an autosomal dominant disorder of connective tissue due to the abnormal expression of fibrillin-1 and is characterized by manifestations in the cardiovascular, musculoskeletal and ophthalmic systems (Table 145.1). The syndrome also shows striking pleiotropism and clinical variability.
Table 145.1 Fibrillin gene disorders
History.
In 1896, the French paediatrician Marfan described a 5-year-old girl with tall stature and disproportionately long limbs and fingers [1]. He used the term ‘dolichostenomelia’ which is now referred to as the marfanoid habitus. A few years later, Marfan’s original patient developed scoliosis [2]. Another clinically similar patient was described by Achard, who introduced the word ‘arachnodactyly’ to describe the associated long, slender fingers [3]. Following reports of associated dislocation of the lens (ectopia lentis) and mitral valve regurgitation with the disorder, Weve, in 1931, proposed the name ‘dystrophica mesodermalis congenita, typus Marfanis’ [4]. This was condensed to Marfan syndrome in 1938 by Apert [5]. In 1956, McKusick, a major contributor to the characterization of MFS, suggested that elastic fibres or a component intimately associated with elastic fibres was defective in MFS. Studies initially focused on collagens, elastin and other connective tissue components and it was only in the late 1980s that fibrillin was identified as a molecule tightly linked with elastic fibres [6,7]. In a very short time, both the positional cloning approach and the candidate gene strategy resulted in the cloning and localization of two fibrillin genes [8–12]. As seen in Table 145.1, mutations in the fibrillin-1 gene (FBN1, located on chromosome 15) were identified in patients with both MFS and autosomal dominant ectopia lentis [13]; mutations in the fibrilin-2 gene (FBN2, on chromosome 5) are linked to the MFS-related disorder called congenital contractural arachnodactyly (CCA) [14].
Finally, two anecdotes in the MFS saga are worth mentioning: it is likely that Marfan’s original patient did not have MFS but rather CCA [15], and there is quite an interest in knowing whether US President Abraham Lincoln was affected by MFS [16,17].
Aetiology.
Fibrillin, an acidic glycoprotein with an estimated molecular mass of 350 kDa, is a major constituent of the 10 nm microfibrils of the extracellular matrix. Its primary structure is characterized by several cysteine-rich motifs, reminiscent of the epidermal growth factor (EGF) peptide module that also has six similarly spaced cysteinyl residues [6,18,19]. The role of fibrillin as the underlying cause of MFS is supported by three independent lines of experimental evidence: firstly, antisera to fibrillin showed a decreased amount of microfibrils in MFS tissue samples [20,21]; secondly, defective synthesis and secretion of fibrillin by dermal fibroblasts were demonstrated in 26 probands with MFS [22]; thirdly, linkage and mutational analysis of several affected kindred confirmed a genetic homogeneity between MFS and the fibrillin gene [23].
Functionally, the 10 nm microfibrils, including fibrillin, serve at least three functions: as a link between elastin and other matrix structures (i.e. the basement membrane at the dermoepidermal junction); as a scaffolding upon which elastin is deposited (i.e. in the tunica media of the aorta); and as a structural component in tissues that do not contain elastin (i.e. the ciliary zonule) [24].
The pathogenic role of fibrillin in MFS was thought to be principally structural, causing a defective connective tissue. Recently, a second pathogenic role was described for fibrillin-1 deficient mice [25], which involves a metabolic pathway since these mice have marked dysregulation of transforming growth factor-β (TGF-β) activation and signalling. This is in keeping with the fact that families with variant MFS syndrome showed mutations on the TGF-β receptor genes.
Genetic analysis provided precise insights into the structural and functional features of fibrillin. Initially, however, such studies failed to define predictable genotype/phenotype correlations. This was highly in keeping with the variable expression of MFS features in affected individuals of the same family and implied that other factors are involved in the development of the clinical phenotype. Indeed, the same mutation (P1148A) was shown in individuals with MFS, isolated ectopia lentis (EL) and the MFS-related but clinically distinct Shprintzen–Goldberg syndrome [26]. The first clinicopathological correlations have now emerged through the analysis of 803 pathogenic mutations found in 1013 probands [27]. These correlations among different mutation types and clinical manifestations might be explained by different underlying genetic mechanisms (dominant-negative versus haploinsufficiency) and by consideration of the two main physiological functions of fibrillin-1 (structural versus as a mediator of TGF-β signalling).
Clinical Features.
The clinical expression of MFS is highly variable, ranging from congenital presentation and death before a few months of life to long-term survival. Severe cases are often sporadic rather than familial and a few have a recessive mode of inheritance with homozygous mutations.
Musculoskeletal Features
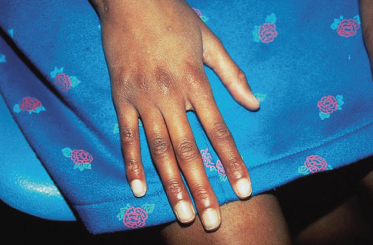
Dolichostenomelia is the characteristic skeletal abnormality in MFS [28]. It includes tall stature, decreased upper to lower segment ratio (US/LS, a value <0.85 in adults is significant), dolichocephaly and arachnodactyly of fingers and toes (Fig. 145.2). Pectus excavatum/carinatum can be present at birth or develop as a result of excessive longitudinal growth. Scoliosis or kyphoscoliosis occurs in 30–60% of MFS individuals. Ligamentous laxity and generalized hypermobility are common and can cause spinal pain, arthralgia, ligament injury or other musculoskeletal symptoms in up to three-quarters of children and in nearly all adults with MFS [29].
Cardiovascular Manifestations
These are responsible for the shortened lifespan of MFS individuals. Multivalvular incompetence predominates in childhood whereas most adults suffer from degenerative complications of the aorta. Dilation of the aorta is due to typical cystic medial necrolysis and provokes aortic aneurysms, often before the age of 40 years. Aortic regurgitation, which correlates with the aortic root diameter, is the most common valvular complication (as high as 70% in adults). Mitral valve prolapse is the next most frequent finding.
Ocular Manifestations
Ectopia lentis (EL) is usually bilateral and occurs in 50–80% of MFS individuals. The lens displacement is usually upward. Although EL is the most frequent ocular manifestation in MFS, visual loss more often results from secondary myopia, retinal detachment, glaucoma or iritis [30–32].
Cutaneous Manifestations
These are minor findings in MFS. Striae atrophicae are the most common cutaneous manifestations of MFS and are usually found on the deltoid, pectoral and lumbar regions [33–35]. They can be found in two-thirds of MFS individuals and their number increases with age [29]. Histologically, they do not differ from striae in the normal population [36].
Skin hyperextensibility is also found in two-thirds of MFS patients, regardless of their age, and significantly correlates with joint laxity [29]. The combination of a marfanoid habitus and skin hyperextensibility, but apparently without MFS, has been described [37].
Easy bruising can be a complaint of MFS patients, this being due not to impaired coagulation but rather as a consequence of thin skin, decreased subcutaneous fat and increased tendency to contusions as a result of joint laxity and poor visual acuity.
Papyraceous scars can occur but are not a typical feature of MFS patients whose cutaneous incisions or lacerations usually heal promptly, in contrast to those with Ehlers–Danlos syndrome [29].
Finally, isolated reports include the association of MFS with poliosis [38], LEOPARD syndrome [39], Ehlers–Danlos syndrome [40], neurofibromatosis [41], PHACE syndrome [42], elastosis perforans serpiginosa [43] and vermiculate atrophoderma [44].
Diagnosis.
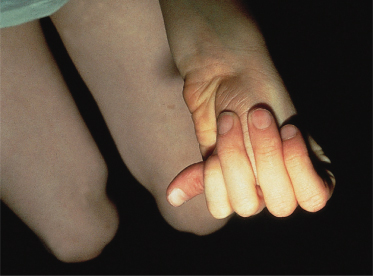
The diagnosis of MFS is mainly clinical [45] and relies on the presence of cardinal manifestations (Box 145.2). Helpful and simple screening tests include the thumb sign (Fig. 145.3), the wrist sign (positive if the thumb and little finger overlap when wrapped around the opposite wrist) and the US/LS ratio. A severe infantile form of the disorder is recognized [46].
Box 145.2 Criteria for the Diagnosis of Marfan Syndrome. Modified from Judge 2005 [59]
Index Case
- If the family/genetic history is not contributory, major criteria in two or more different organ systems and involvement of a third organ system.
- If a mutation known to cause Marfan syndrome in others is identified, one major criterion in an organ system and involvement of a second organ system.
Relative of an Index Case Who Independently Meets these Strict Diagnostic Criteria
Presence of a major criterion in the family history, one major criterion in an organ system, and involvement of a second organ system.
Criteria
Skeletal System
A major criterion requires at least four of the following: pectus carinatum; pectus excavatum needing surgery; reduced upper-to-lower segment ratio or arm span-to-height ratio ≥1.05; positive wrist and thumb signs; reduced extension of the elbows (≤170°); medial displacement of the medial malleolus causing pes planus; protrusio acetabulae of any degree.
Involvement is defined by the presence of two of the preceding features or the presence of one of the preceding features and two of the following minor criteria: pectus excavatum not requiring surgery; joint hypermobility; high-arched palate with crowding of teeth; facial appearance (dolichocephaly, malar hypoplasia, enophthalmos, retrognathia, down-slanting palpebral fissures).
Ocular System
A major criterion is defined by ectopia lentis.
Involvement is defined by the presence of at least two of the following minor criteria: flat cornea; increased axial length of globe; hypoplastic iris or hypoplastic ciliary muscle causing decreased miosis.
Cardiovascular System
A major criterion is defined by either dissection of the ascending aorta or dilation of the ascending aorta, with or without aortic regurgitation, and involving at least the sinuses of Valsalva.
Involvement requires the presence of at least one of the following minor criteria: mitral valve prolapse with or without mitral regurgitation; dilation of the main pulmonary artery, in the absence of valvular or peripheral pulmonic stenosis, under the age of 40 years; calcification of the mitral annulus under the age of 40 years; dilation or dissection of the descending thoracic or abdominal aorta under the age of 50 years.
Pulmonary System
No major criterion.
Involvement is defined by at least one of the following minor criteria: spontaneous pneumothorax; radiological evidence of apical blebs.
Skin and Integument
No major criterion.
Involvement is defined by at least one of the following minor criteria: striae atrophicae; a recurrent or incisional hernia.
Dura
A major criterion is the presence of lumbosacral dural ectasia.
No minor criterion.
Family/genetic history
A major criterion is defined by one of the following: having a parent, child or sibling who meets these diagnostic criteria independently; presence of a mutation of FBN1 which is known to cause Marfan syndrome; presence of a haplotype around the FBN1 locus, inherited by descent, known to be associated with unequivocally diagnosed Marfan syndrome in the family.
No minor criterion.
A marfanoid habitus is found in two rarer fibrillin disorders (see Table 145.1). Congenital contractural arachnodactyly (CCA) presents with joint contractures and abnormalities of the external ears but without cardiovascular or ocular manifestations [14,15]. The marfanoid-craniosynostosis syndrome (Shprintzen–Goldberg) is a clinically distinct variant of MFS, reported in only 11 individuals and characterized by additional findings such as craniofacial abnormalities, mental retardation, hypotonia and congenital abdominal wall weakness [27,47].
Other differential diagnoses include homocystinuria, Stickler syndrome (hereditary arthrophthalmopathy), annuloaortic ectasia (Erdheim disease) and mitral valve prolapse syndrome [48,49].
Management.
The dermatologist will rarely need to be involved in multidisciplinary teams managing MFS. In recent years, the increased lifespan and decreased morbidity in MFS have mainly been achieved through the improvement of cardiovascular, orthopaedic and ocular surgery [32,50–52]. β-blockers are useful to reduce/delay aortic dilation [53,54]. Pregnancy remains a problem, due to the high risk of ruptured aneurysms, but can be managed in women with minimal cardiovascular findings [55–57].
A breakthrough in therapy came from the finding that selected manifestations of MFS reflect excessive signalling by the TGF-β family of cytokines. Promising results have been seen with angiotensin II-receptor blockers on aortic root dilation [58].
References
1 Marfan A. Un cas de déformation congénitale des quatre membres plus prononcée aux extrémités, charactérisée par l’allongement des os avec un certain degré d’amincissement. Bull Mem Soc Méd Hôp Paris 1896;13:220.
2 Méry H, Babonneix L. Un cas de déformation congénitale des quatre membres: hyperchondroplasie. Bull Mem Soc Méd Hôp Paris 1902;19:671.
3 Achard C. Arachnodactylie. Bull Mem Soc Méd Hôp Paris 1902;19:834.
4 Weve H. Veber Arachnodactylie (Dystrophia mesodermalis congenita, typus Marfanis). Arch Augenheilk 1931;104:1.
5 Apert E. (Les formes frustes du syndrome dolichosténomélique de Marfan. Nourisson 1938;26:1.
6 Sakai L, Keene D, Engvall E. ( Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol 1986;103:2499.
7 Sakai LY, Keene DR. Fibrillin: monomers and microfibrils. Methods Enzymol 1994;245:29–52.
8 Blanton S, Sarfarazi M, Eiberg H et al. (An exclusion map of Marfan syndrome. J Med Genet 1990;27:73–7.
9 Dietz H, Pyeritz R, Hall B et al. The Marfan syndrome locus: confirmation of assignment to chromosome 15 and identification of tightly linked markers at 15q15–q21.3. Genomics 1991;9:355–61.
10 Dietz H, Cutting G, Pyeritz R et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene see comments. Nature 1991;352:337–9.
11 Lee B, Godfrey M, Vitale E et al. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes see comments. Nature 1991;352:330–4.
12 Maslen C, Corson G, Maddox B et al. Partial sequence of a candidate gene for the Marfan syndrome see comments. Nature 1991;352:334–7.
13 Kainulainen K, Karttunen L, Puhakka L et al. Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome. Nat Genet 1994;6:64–9.
14 Putnam E, Zhang H, Ramirez F et al. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat Genet 1995;11:456–8.
15 Beals R, Hecht F. Congenital contractural arachnodactyly. A heritable disorder of connective tissue. J Bone Joint Surg Am 1971;53:987–93.
16 Gordon A. Abraham Lincoln, the famous case of Marfan’s syndrome. Deutsch Med J 1967;18:256–60.
17 McKusick V. The defect in Marfan syndrome. Nature 1991;352:279–81.
18 Pereira L, d’Alessio M, Ramirez F et al. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum Mol Genet 1993;2:1762.
19 Handford P, Downing A, Rao Z et al. The calcium binding properties and molecular organization of epidermal growth factor-like domains in human fibrillin-1. J Biol Chem 1995;270:6751–6.
20 Hollister D, Godfrey M, Sakai L et al. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med 1990;323:152–9.
21 Godfrey M, Menashe V, Weleber R et al. Cosegregation of elastin-associated microfibrillar abnormalities with the Marfan phenotype in families. Am J Hum Genet 1990;46:652–60.
22 Milewicz D, Pyeritz R, Crawford E et al. Marfan syndrome: defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J Clin Invest 1992;89:79–86.
23 Tsipouras P, del Mastro R, Sarfarazi M et al. Genetic linkage of the Marfan syndrome, ectopia lentis, and congenital contractural arachnodactyly to the fibrillin genes on chromosomes 15 and 5. The International Marfan Syndrome Collaborative Study. N Engl J Med 1992;326:905–9.
24 Godfrey M. From fluorescence to the gene: the skin in the Marfan syndrome. J Invest Dermatol 1994;103:58S–62S.
25 Neptune ER, Frischmeyer PA, Arking DE. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003;33:407–11.
26 Sood S, Eldadah Z, Krause W et al. Mutation in fibrillin-1 and the Marfanoid–craniosynostosis (Shprintzen–Goldberg) syndrome. Nat Genet 1996;12:209–11.
27 Faivre L, Collod-Beroud G, Loeys BL et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 2007;81: 454–66.
28 Pyeritz R, McKusick V. The Marfan syndrome: diagnosis and management. N Engl J Med 1979;300:772–7.
29 Grahame R, Pyeritz R. The Marfan syndrome: joint and skin manifestations are prevalent and correlated. Br J Rheumatol 1995;34:126–31.
30 Maumenee I. The eye in the Marfan syndrome. Trans Am Ophthalmol Soc 1981;79:684–733.
31 Nelson L, Maumenee I. Ectopia lentis. Surv Ophthalmol 1982;27:143–60.
32 Roussat B, Chiou A, Quesnot S et al. Surgery of ectopia lentis in Marfan disease in children and young adults. J Fr Ophtalmol 1995;18:170–7.
33 Fazekas A, Szego L, Vigvary L. Striae distensae elasticae in Marfan’s syndrome. Orv Hetil 1973;114:2290–2.
34 McKusick V. Transverse striae distensae in the lumbar area in father and two sons. Birth Defects 1971;7:260–1.
35 Shuster S. The cause of striae distensae. Acta Derm Venereol Suppl Stockh 1979;59:161–9.
36 Pinkus H, Keech M, Mehregan A. Histopathology of striae distensae, with special reference to striae and wound healing in the Marfan syndrome. J Invest Dermatol 1966;46:283–92.
37 Sakatsume Y, Saito M, Hara Y et al. A case of marfanoid body habitus associated with an excessive hyperextensibility of the skin – an unclassified case in inherited connective tissue diseases. Nippon Naika Gakkai Zasshi 1988;77:499–505.
38 Herman K, Salman K, Rose L. ( White forelock in Marfan’s syndrome: an unusual association, with review of the literature. Cutis 1991;48:82–4.
39 Torok L, Szentendrei L, Szili M et al. Progressive cardiomyopathic lentiginosis (LEOPARD syndrome) in 3 patients, combined with Marfan syndrome. Z Hautkr 1990;65:197–201.
40 Fazekas A. Simultaneous occurrence of Ehlers–Danlos syndrome and Marfan’s syndrome. Orv Hetil 1976;117:154–8.
41 Copeland T, Tiwary C, Coker S. Coexistence of neurofibromatosis and Marfan’s syndrome. South Med J 1986;79:489–92.
42 Slavotinek A, Dubovsky E, Dietz H et al. Report of a child with aortic aneurysm, orofacial clefting, hemangioma, upper sternal defect, and marfanoid features: possible PHACE syndrome. Am J Med Genet 2002;110:283–8.
43 Mehregan A. Elastosis perforans serpiginosa: a review of the literature and report of 11 cases. Arch Dermatol 1968;97:381–93.
44 Harper J, Sidwell R. Vermiculate atrophoderma in a boy with Marfan syndrome. Br J Dermatol 1999;141:750–2.
45 Beighton P, de Paepe A, Danks D et al. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet 1988;29:581–94.
46 Morse R, Rockenmacher S, Pyeritz R et al. Diagnosis and management of infantile marfan syndrome. Pediatrics 1990;86:888–95.
47 Shprintzen R, Goldberg R. A recurrent pattern syndrome of craniosynostosis associated with arachnodactyly and abdominal hernias. J Craniofac Genet Dev Biol 1982;2:65–74.
48 Devereux R, Brown W. Genetics of mitral valve prolapse. Prog Med Genet 1983;5:139–61.
49 Glesby M, Pyeritz R. Association of mitral valve prolapse and systemic abnormalities of connective tissue. A phenotypic continuum see comments. JAMA 1989;262:523–8.
50 Sponseller P, Hobbs W, Riley L et al. The thoracolumbar spine in Marfan syndrome. J Bone Joint Surg Am 1995;77:867–76.
51 Gott V, Gillinov A, Pyeritz R et al. Aortic root replacement. Risk factor analysis of a seventeen-year experience with 270 patients. J Thorac Cardiovasc Surg
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


