AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive.
Cutaneous Aspects
Cutaneous features of EDS include skin hyperextensibility and fragility [4,6,55]. The skin is soft, and is often described as velvety and doughy with a chamois leather texture. The skin is easily extended and, upon release, recoils back to its original shape (Fig. 142.1). Redundant skin may be present overlying elbows and knees, but this differs from the lax skin of cutis laxa which, after stretching, does not recoil. Hyperextensibility and skin softness can be difficult to assess in infants because of subcutaneous fat and are more easily evaluated in slightly older children.
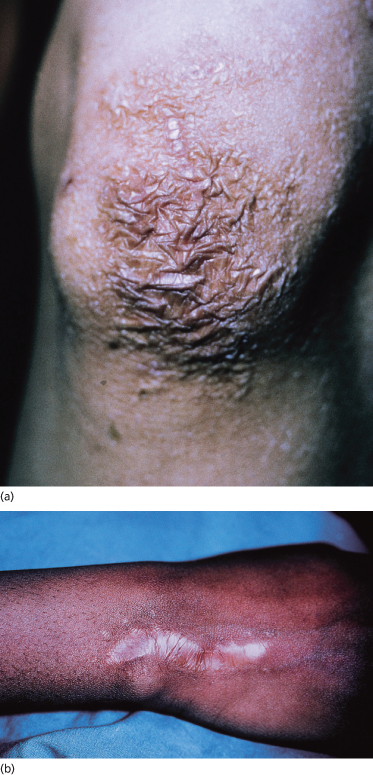
Skin fragility (dermatorrhexis) is manifested by tearing of the skin after minor trauma. Sites commonly involved include knees, shins, elbows and face, especially the forehead and chin (Fig. 142.2). Scarring occurs most commonly during childhood as infants begin to crawl and walk and experience minor trauma, and improves with increasing age. Cutaneous fragility also results in easy bruising, delayed wound healing and poor healing after suturing, with a higher incidence of wound dehiscence. Prominent bruising can lead to mistaken concerns about child abuse in affected children. Scars characteristically widen and develop fine ‘cigarette paper’ or ‘papyraceous’ wrinkling in addition to a shiny surface with overlying telangiectasias.
Fig. 142.2 Skin fragility with abnormal scarring. (a) Knee with cigarette paper or papyraceous wrinkling after trauma. (b) Scar widening after healing.
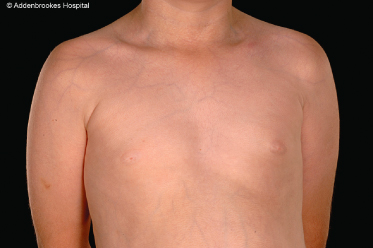
Vascular EDS typically lacks significant hyperextensibility of skin but is associated with pale skin with readily visible veins on the torso, particularly the upper chest (Fig. 142.3) and dorsum of hands and feet (acrogeric appearance).
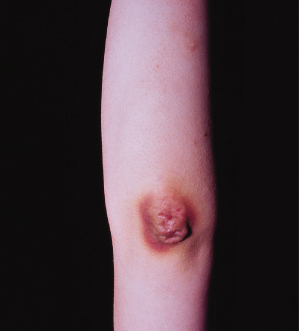
Unique cutaneous findings in some patients with EDS are molluscoid pseudo-tumours (Fig. 142.4) and spheroids. Molluscoid pseudo-tumours are fibrous nodules that occur over areas of recurrent trauma such as the elbows, knees and heels. Spheroids are small subcutaneous, cyst-like nodules that occur over bony prominences of the legs and arms. These are calcified on radiography examination and result from trauma-induced fibrosis and calcification of the subcutaneous fat lobules [56,57].
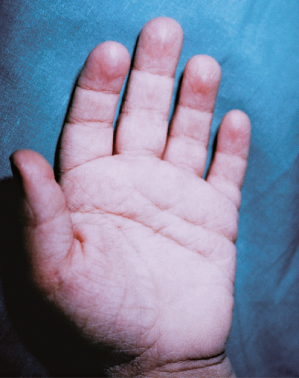
Additional cutaneous features include looseness of palmar skin and an increased number of palmar creases (Fig. 142.5). Elastosis perforans serpiginosa has been reported in some affected individuals [58,59] and is more common in the vascular subtype. Piezogenic papules, herniations of fat lobules through fascia at the medial and lateral aspects of the feet, may also be present. Pregnancy-associated striae gravidarum generally do not occur in patients with EDS.
Musculoskeletal Aspects
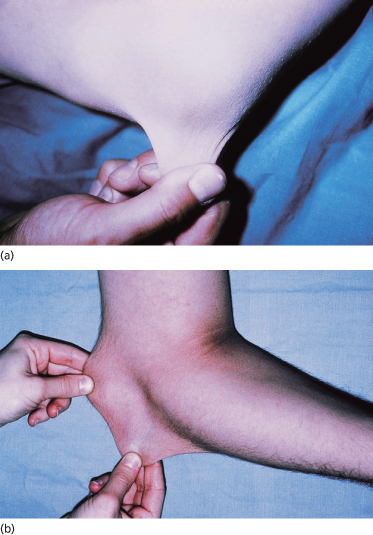
With the exception of vascular EDS, in which only small joints are involved, joint hypermobility is a cardinal feature of EDS and involves both large and small joints (Fig. 142.6) [60]. Generally, this is first noticed when a child exhibits a delay in beginning to walk and remains unsteady with a greater number of falls and improves with increasing age. At any age, females display greater mobility than males. Clinical evaluation of joint hyperextensibility can be made using the Beighton score [61] (Box 142.1). This includes examination of small joints by assessing the ability to dorsiflex the fifth finger beyond 90° and the ability to appose thumb to flexor forearm. Large joint hyperextensibility is evaluated by the presence of knee and elbow hyperextension. One point is given for each side of the body for these manoeuvres and a further point is added for the ability to bend and touch palms of hands flat to the floor. Hypermobility is present if a score of four out of nine or greater is achieved. Severe hypermobility as seen with kyphoscoliosis and arthrochalasia types can be present at birth with hypotonia, skeletal deformities and congenital hip dislocations.
Box 142.1 Beighton scoring system
| Movement | Score |
| Dorsiflexion of right and left 5th finger >90o | 2 |
| Apposition of right and left thumb to forearm | 2 |
| Hyperextension of right and left elbow >10o | 2 |
| Hyperextension of right and left knee >10o | 2 |
| Palms touching the floor with knees extended | 1 |
| Total | 9 |
Reproduced from Beighton et al. Ann Rheum Dis 1973;32:413–18 with permission from BMJ Publishing Group Ltd.
Fig. 142.6 Joint hypermobility. (a,b) Small joint hypermobility with extension of fingers beyond 90°. (c) Large joint hypermobility is evidenced by hyperextension of the knees.
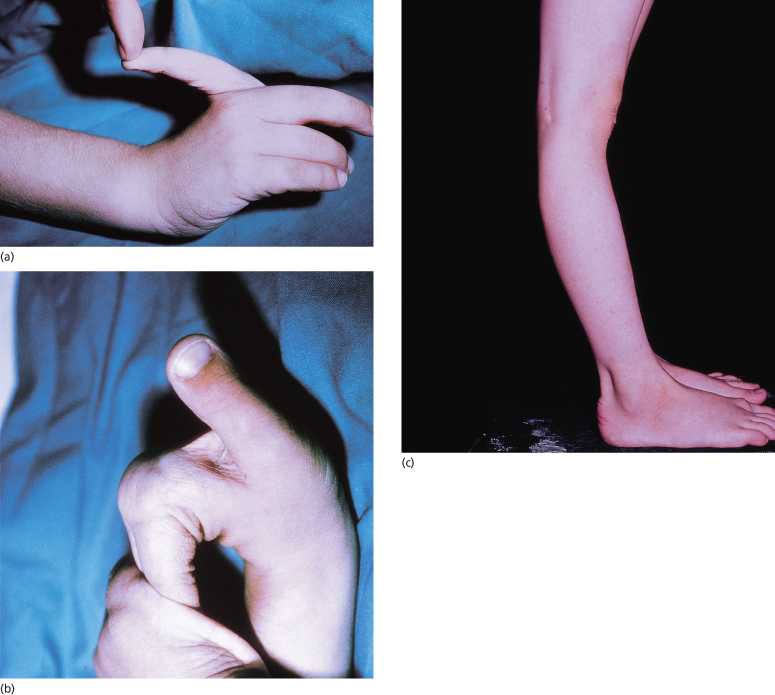
Early-onset osteo-arthritis can be a significant complication and usually involves weight-bearing joints such as the knees and ankles. Chronic pain is a common manifestation of EDS [62]. Other musculoskeletal complications include recurrent joint effusions, which occur most commonly in knees, ankles and elbows, pes planus (flat-footedness) and kyphoscoliosis [63]. Haemarthrosis may occur in vascular EDS. Joint instability results in delayed walking in childhood and joint subluxation. Children and adults with hypermobility are more prone to reduced bone mineral density and DEXA screening has been advocated for this group [64,65].
Cardiovascular Aspects
Structural cardiac abnormalities are not specifically associated with EDS and may be present as a result of random association [66,67]. Mitral valve prolapse (MVP), resulting from redundant chordae tendineae and valve cusps, has been reported, most often with hypermobile and vascular EDS [68]; however, one study suggests that the incidence of MVP in these types and classic EDS is not increased [69]. A rare, recessively inherited form of EDS due to heterozygous mutations in COL1A2, characterized by cardinal features, is also associated with cardiac valvular defects which may not present until adult life [24,70]. Vascular EDS is also associated with spontaneous and life-threatening rupture of large arteries, especially the descending aorta and other abdominal vessels [71]. Intracranial aneurysm and arteriovenous fistulae rupture may occur with vascular EDS, but have also been reported with other EDS types [72].
Genitourinary Aspects
Urinary bladder diverticulae are found in most EDS types but are specifically associated with occipital horn syndrome (formerly EDS IX). These may result in vesicoureteral reflex and have an associated risk of recurrent urinary tract infections.
Neurological Aspects
Autonomic nervous system-related symptoms (dysautonomia) such as syncope, palpitations, easy fatigue and postural orthostatic intolerance (POTS) occur more commonly in hypermobile patients [73]. A small subset also reports the failure of intradermal local anaesthetic [74]. Significant neurological complications include muscle hypotonia in kyphoscoliotic EDS and haemorrhage from intracranial arterial aneurysm in vascular EDS. Joint laxity, particularly of the spine, may result in nerve trauma and subsequent neurological manifestations [75,76].
Ocular Aspects
Minor ocular findings include epicanthal folds, ease in everting the upper eyelid (Méténier’s sign), redundant upper eyelid skin and strabismus that is secondary to the laxity of tendons supporting the extrinsic muscles of the eyes [77]. Kyphoscoliosis-type EDS (also formerly known as ocular-scoliotic variant) has been associated with more significant ocular complications including retinal detachment and scleral perforation, which may occur during childhood [78]. Microcornea, glaucoma and keratoconus may also occur. However, a more recent review of patients suggests that the more serious ocular complications have been overestimated and that myopia is the most common finding in this EDS subgroup [79]. Keratoconus may be present in classic EDS and has been described co-segregating in a family with EDS II found to have a COL5A2 mutation [80].
Dental and Oral Aspects
Gorlin’s sign, the ability to touch the tip of the nose with the tongue, has sometimes been associated with EDS; nonetheless, this finding is not specific as it occurs in 10% of unaffected individuals [81]. Periodontitis-type EDS is associated with early-onset periodontal disease and premature loss of teeth [82]. It has been suggested that absence of the inferior labial and lingual frenula may be a reliable marker for classic and hypermobile EDS [83], although this has been disputed by other authors [84,85].
Bleeding Disorders
Bleeding and bruising problems are seen in different forms of EDS, particularly in the vascular EDS subtype. In children with EDS, excessive bruising is often the presenting problem and a careful diagnosis is necessary to distinguish between this disorder and other potential causes of bruising [86].
Obstetric Complications
Complications of pregnancy occur in both mother and infant with EDS and are related to the specific type [87]. These include maternal joint subluxation, severe varicose veins and cervical insufficiency, with risk of miscarriage and preterm labour and postpartum haemorrhage. Vascular EDS is associated with the greatest risks, including uterine rupture, which has an estimated maternal mortality of 25%. Fetuses affected with EDS have a higher incidence of prematurity, resulting from maternal premature rupture of membranes, in addition to demonstrating low birthweight and short stature.
Prognosis.
The prognosis of the various EDS subtypes is based upon the specific associated features and their complications and has been reviewed above.
Differential Diagnosis.
The differential diagnosis of EDS includes syndromes specifically associated with joint hypermobility, cutaneous laxity or aneurysms. Familial articular hypermobility syndrome was initially classified as EDS XI but is manifest by joint laxity alone, without any skin changes, and must be considered in patients with joint hypermobility without other stigmata of EDS. Cutis laxa has increased cutaneous laxity with acrogeric appearance but is distinguished by lack of recoil after stretching of the skin.
Occasional patients with hypermobility-type and kyphoscoliosis-type EDS have a marfanoid habitus, suggesting some overlap with Marfan syndrome [88–90].
Loeys–Dietz syndrome is a recently described aneursymal phenotype due to mutations in transforming growth factor β receptors 1 and 2 (TGFBR 1 or 2) [91]. Patients with type I LDS have overlapping features with Marfan syndrome, most noticeably aortic aneurysms, arachnodactyly and dural ectasia. Additional features of cleft palate or bifid uvula, craniosynostosis and hypertelorism occur. Type II LDS phenotype overlaps with vascular EDS although a bifid uvula may rarely be present. Although the overall prognosis is poor for both LDS and vascular EDS, the survival during, or immediately after, vascular surgery is dramatically lower in the former.
The occipital horn syndrome is a rare disorder with X-linked recessive inheritance. It arises from mutations in the same gene as for Menkes syndrome [92] and has been reclassified as a disorder of copper transport. It is characterized, at birth, by soft, lax and redundant skin. The skin, however, is not hyperelastic and does not demonstrate easy bruising or abnormal scarring. During childhood, formation of bladder diverticuli may occur and skeletal changes such as exostoses at sites of tendon insertion, also called occipital horns, are typical.
Arthrochalasia-type EDS can also share features with osteogenesis imperfecta [93] and a non-glycine substitution has been recently reported in the α1(I)collagen chain in a family with an osteogenesis imperfecta/EDS phenotype [94].
Investigations.
The diagnosis of EDS is based primarily on clinical history, family history to clarify inheritance and careful physical examination to evaluate involvement of the various systems. Biochemical testing may be undertaken to confirm the diagnosis in several EDS subtypes. In vascular EDS, protein chemistry analysis using fibroblast cultures demonstrates decreased synthesis or secretion of procollagen III. In addition, decreased serum levels of procollagen III aminopeptide may be found [30]. If collagen III levels are normal, type II LDS should be considered and TGFBR1 and 2 genes should be screened for mutations [91].
Decreased lysyl hydroxylase enzyme activity in fibroblast culture resulting in underhydroxylated collagen causes abnormally fast migration of type I collagen on radiolabelled protein electrophoresis and confirms the diagnosis of kyphoscoliosis-type EDS [35]. Measurement of urinary excretion of lysyl- and hydroxylysyl-derived pyridinoline cross-links is a non-invasive reliable diagnostic test in this subgroup [34]. Arthrochalasia subtypes can be confirmed through evaluation of type I collagen processing, which shows abnormalities in α1, α2 or both peptides [38,39,42]. Electron microscopic studies of skin in dermatosparaxis show markedly irregular (hieroglyphic-like) collagen fibrils. Biochemical confirmation is based on electrophoretic analysis showing the accumulation of pNα1(I) and pNα2(I) chains of type I collagen extracted from dermis [44].
Tenascin-X deficient EDS is confirmed with the complete absence of serum TNX, measured by ELISA [50]. Haploinsufficiency associated with hypermobility EDS results in reduced serum TNX levels [22].
The genetic defects have been elucidated for vascular, kyphoscoliosis, arthochalasia, dermatosparaxis, tenascin-X deficient and some classic subtypes, and molecular analysis is therefore feasible, but it is usually available only in specialized centres.
Treatment.
The management of an EDS patient is dependent upon which system is involved. Joint laxity may result in joint dislocations or early-onset osteo-arthritis and may require orthopaedic or rheumatological follow-up. Physical therapy to strengthen supporting muscles and stabilize loose joints may be necessary in children who are experiencing delay in motor development. Bracing may be required to support unstable joints and assist with walking. Sports with joint impact such as gymnastics or long-distance running are best avoided in children with significant joint hyperextensibility. Repeated hyperextension of joints to perform ‘joint tricks’ should also be discouraged as this stresses an unstable joint, increasing the risk of joint dislocation. In children with severe skin fragility, protective pads over knees, shins and elbows may be helpful in preventing lacerations. Poor wound healing and scar formation have implications for any surgical procedure to be performed, and the surgeon must be aware of the potential problems.
Because of the risk of sudden, and potentially fatal, vascular or bowel haemorrhage in vascular EDS, patients should be advised to wear a medical alert tag or bracelet with their diagnosis. They should be given advice to avoid any activities that are likely to cause a sudden increase in blood pressure. Females with vascular EDS must be made aware of the complications associated with pregnancy. This also applies to females with the kyphoscoliosis type.
Most importantly, counselling, reviewing the inheritance and specific features of the subtype involved, is essential for all families. The clinical and genetic heterogeneity of the EDS subtypes must be stressed with careful explanation of the expected risks and complications associated with the specific diagnosis. Clearly, the diagnosis of mild classic EDS has minimal impact on family planning or activity level of an affected individual, whereas the diagnosis of vascular, kyphoscoliosis, arthrochalasia or dermatosparaxis types has significant lifelong sequelae. The dramatic arterial complications of vascular-type EDS have often been stressed in the older literature on EDS, and it is essential to clarify that these are not associated with all the EDS subtypes. Patient support groups exist in the UK (www.ehlersdanlos.org) and the USA (www.ednf.org) and are a valuable source of information for patients.
References
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


