* Junctional epidermolysis bullosa is a heterogeneous condition. Other subtypes of this disorder may result from mutations in the gene encoding type XVII collagen (COL17A1/BPAG2) on 10q24.3 [30].
† Junctional epidermolysis bullosa with pyloric atresia may also be caused by mutations in the gene for α6 integrin (ITGA6) on 2q31.1 [31].
‡ Some cases of epidermolysis bullosa simplex are caused by mutations in the keratin 5 gene (KRT5) on 12q13.13 [32].
§ Some cases of bullous congenital ichthyosiform erythroderma arise because of mutations in the gene encoding keratin 1 (KRT1) on 12q13.13 [33].
** Some cases of lamellar ichthyosis show linkage to different loci on 2q33–35 [34], 3p21 and 19p12–q12 [35].
Fig. 139.1 Chorionic villi sampled at 11 weeks’ gestation. These villi have had all traces of maternal decidua carefully removed so that pure fetal DNA can subsequently be extracted without the risk of maternal DNA contamination. The cleaned villi are digested using proteinase K or urea buffers, DNA is then precipitated following which previously optimized and characterized PCT protocols can be used to test for the presence or absence of mutations in a DNA-based prenatal test.

Apart from CVS, fetal DNA can also be extracted from fetal cells obtained by amniocentesis; this is usually performed at about 16 weeks’ gestation [37]. Amniotic fluid and its cells can be examined for morphological, cytogenetic, biochemical or molecular abnormalities. Amniotic fluid cells are derived from fetal epidermis, alimentary and genitourinary mucosa, and amnion. These cells have been used to assess inherited disorders with abnormal DNA synthesis and repair, as well as metabolic disorders that result in abnormal protein synthesis. However, now that the molecular basis of nearly all these conditions is known, such approaches are rarely necessary and most prenatal tests can be performed by direct analysis of genomic DNA pathology in fetal DNA following CVS or amniocentesis [38].
A number of inherited skin disorders in which specific gene mutations have been identified and used as a basis for first-trimester DNA-based prenatal diagnosis have been documented and are shown in Table 139.1, and an example of DNA analysis for one of these conditions is illustrated in Figure 139.2. To these disorders, several further conditions might be added in the near future, based on recent disclosures of pathogenic molecular events. Over the last 15 years, the most common dermatological indications for DNA-based prenatal testing have been Hallopeau–Siemens recessive dystrophic epidermolysis bullosa (EB) and Herlitz junctional EB [38]. Inherited disorders such as lamellar ichthyosis are also suitable for DNA-based prenatal testing [12], but this form of ichthyosis shows considerable genetic heterogeneity [34,35]. Therefore it is vital that specific abnormalities should first be identified in the proband, such as mutations in the keratinocyte transglutaminase gene (TGM1 on 14q11.2), before any prenatal test is planned. Clues to inherent TGM1 gene pathology in an individual case of lamellar ichthyosis can be obtained from immunohistochemical studies for the TGM1 enzyme in the proband’s skin [39,40]. Likewise, junctional EB is a heterogeneous disorder that results from autosomal recessive mutations in six different genes encoding structural components of the hemi-desmosomal anchoring filament network [41]. Therefore, determining the correct gene responsible for a specific case is crucial to the feasibility and accuracy of DNA-based prenatal testing. To that end, the development of monoclonal antibodies to candidate gene proteins and immunostaining of skin sections from affected individuals have become an integral part of the work-up for molecular studies for several recessive disorders that stem from loss-of-function mutations leading to reduced protein expression in the skin [41,42].
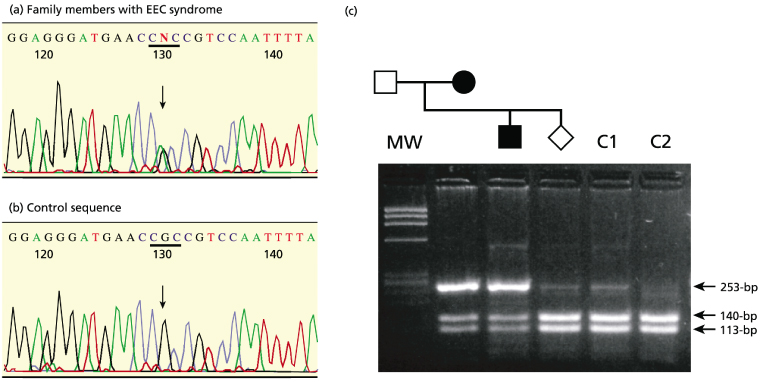
Fig. 139.2 DNA-based prenatal testing. Illustrated is a DNA-based prenatal test for the autosomal dominant ectrodactyly ectodermal dysplasia-clefting (EEC) syndrome. The mother had EEC syndrome, which means that the fetus had a 50% risk of also being affected. She had already had one son with EEC syndrome. (a) Sequencing of amplified DNA from the mother and the previously affected child shows a heterozygous G > A transition in exon 7 of the p63 gene, which changes an arginine residue (CGC) to histidine (CAC). The mutation is designated p.R279H. (b) Sequencing of amplified control DNA for this exon shows only the wild-type arginine codon (CGC). (c) DNA-based prenatal testing using restriction endonuclease digestion with AciI. The mutation p.R279H results in loss of a cut site for AciI. In control DNA (lanes C1 and C2), the PCR product is completely digested into fragments of 140 and 113 bp. By contrast, there is an additional undigested band of 253 bp on DNA from the affected mother and child. The fetal PCR products are digested similarly to the control DNA, indicating that the fetus has inherited two wild-type p63 alleles with respect to this mutation and is therefore predicted to be clinically unaffected with EEC syndrome.
Adapted from South et al. 2002 [18].
References
1 McGrath JA, Kivirikko S, Ciatti S et al. A homozygous nonsense mutation in the alpha 3 chain gene of laminin 5 (LAMA3) in Herlitz junctional epidermolysis bullosa: prenatal exclusion in a fetus at risk. Genomics 1995;29:282–4.
2 Vailly J, Pulkkinen L, Miquel C et al. Identification of a homozygous one-base pair deletion in exon 14 of the LAMB3 gene in a patient with Herlitz junctional epidermolysis bullosa and prenatal diagnosis in a family at risk for recurrence. J Invest Dermatol 1995;104:462–6.
3 Christiano AM, Pulkkinen L, McGrath JA et al. Mutation based prenatal diagnosis of Herlitz junctional epidermolysis bullosa. Prenat Diagn 1997;17:343–54.
4 Ashton GH, Sorelli P, Mellerio JE et al. Alpha 6 beta 4 integrin abnormalities in junctional epidermolysis bullosa with pyloric atresia. Br J Dermatol 2001;144:408–14.
5 Hovnanian A, Hilal L, Blanchet-Bardon C et al. DNA-based prenatal diagnosis of generalized recessive dystrophic epidermolysis bullosa in six pregnancies at risk for recurrence. J Invest Dermatol 1995;104:456–61.
6 Christiano AM, LaForgia S, Paller AS et al. Prenatal diagnosis for recessive dystrophic epidermolysis bullosa in 10 families by mutation and haplotype analysis in the type VII collagen gene (COL7A1). Mol Med 1996;2:59–76.
7 McGrath JA, Dunnill MG, Christiano AM et al. First trimester DNA-based exclusion of recessive dystrophic epidermolysis bullosa from chorionic villus sampling. Br J Dermatol 1996;134:734–9.
8 Klinberg S, Mortimore R, Parkes J et al. Prenatal diagnosis of dominant dystrophic epidermolysis bullosa by COL7A1 molecular analysis. Prenat Diagn 2000;20:618–22.
9 Rugg EL, Baty D, Shemanko CS et al. DNA-based prenatal testing for the skin blistering disorder epidermolysis bullosa simplex. Prenat Diagn 2000;20:371–7.
10 Rothnagel JA, Longley NA, Holder RA et al. Prenatal diagnosis of epidermolytic hyperkeratosis by direct gene sequencing. J Invest Dermatol 1994;102:13–16.
11 Sprecher E, Chavanas S, DiGiovanna JJ et al. The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: implications for mutation detection and first case of prenatal diagnosis. J Invest Dermatol 2001;117:179–87.
12 Schorderet DF, Huber M, Laurini RN et al. Prenatal diagnosis of lamellar ichthyosis by direct mutational analysis of the keratinocyte transglutaminase gene. Prenat Diagn 1997;17:483–6.
13 Sillen A, Holmgren G, Wadelius C. First prenatal diagnosis by mutation analysis in a family with Sjogren–Larsson syndrome. Prenat Diagn 1997;17:1147–9.
14 Yeowell HN, Walker LC, Farmer B et al. Mutational analysis of the lysyl hydroxylase I gene (PLOD) in six unrelated patients with Ehlers–Danlos syndrome type VI: prenatal exclusion of this disorder in one family. Hum Mutat 2000;16:90.
15 Shimizu H, Niizeki H, Suzumori K et al. Prenatal diagnosis of oculocutaneous albinism by analysis of the fetal tyrosinase gene. J Invest Dermatol 1994;103:104–6.
16 Falik-Borenstein TC, Holmes SA, Borochowitz Z et al. DNA-based carrier detection and prenatal diagnosis of tyrosinase-negative oculocutaneous albinism (OCA1A). Prenat Diagn 1995;15:345–9.
17 Daika-Dahmane F, Dommergues M, Narcy F et al. Congenital erythropoietic porphyria: prenatal diagnosis and autopsy findings in two sibling fetuses. Pediatr Dev Pathol 2001;4:180–4.
18 South AP, Ashton GH, Willoughby C et al. EEC syndrome: heterozygous mutation in the p63 gene and DNA-based prenatal diagnosis. Br J Dermatol 2002;146:216–20.
19 Nowaczyk MJ, Garcia DM, Eng B et al. Rapid molecular prenatal diagnosis of Smith–Lemli–Opitz syndrome. Am J Med Genet 2001;102:387–8.
20 Bunge S, Steglich C, Lorenz P et al. Prenatal diagnosis and carrier detection in mucopolysaccharidosis type II by mutation analysis. A 47,XXY male heterozygous for a missense point mutation. Prenat Diagn 1994;14:777–80.
21 Sergi C, Penzel R, Uhl J et al. Prenatal diagnosis and fetal pathology in a Turkish family harbouring a novel nonsense mutation in the lysosomal alpha-N-acetyl-neuraminidase (sialidase) gene. Hum Genet 2001;109:421–8.
22 De Vries BB, Kleijer WJ, Keulemans JL et al. First trimester diagnosis of infantile neuronal ceroid lipofuscionosis (INCL) using PPT enzyme assay and CLN1 mutation analysis. Prenat Diagn 1999;19:559–62.
23 Kleijer WJ, van Diggelen OP, Keulemans JL et al. First trimester diagnosis of late-infantile neuronal ceroid lipofuscionosis (LINCL) by tripeptidyl peptidase I assay and CLN2 mutation analysis. Prenat Diagn 2001;21:99–101.
24 Chen CP, Chern SR, Shih JC et al. Prenatal diagnosis and genetic analysis of type I and type II thanatophoric dysplasia. Prenat Diagn 2001;21:89–95.
25 Chitayat D, Fernandez B, Gardner A et al. Compound heterozygosity for the achondroplasia-hypochondroplasia FGFR3 mutations: prenatal diagnosis and postnatal outcome. Am J Hum Genet 1999;84:401–5.
26 Milunsky JM, Maher TA, Metzenberg AB. Molecular, biochemical and phenotypic analysis of a hemizygous male with a severe atypical phenotype for X-linked dominant Conradi–Hunermann–Happle syndrome and a mutation in EBP. Am J Hum Genet 2003;116:249–54.
27 Yang Y, Ding B, Wang K et al. DNA-based prenatal diagnosis in a Chinese family with xeroderma pigmentosum group A. Br J Dermatol 2004;150:1190–3.
28 Maydan G, Andresen BS, Madsen PP et al. TAT gene mutation analysis in three Palestinian kindreds with oculocutaneous tyrosinaemia type II: characterization of a silent exonic transversion that causes missplicing by exon 11 skipping. J Inherit Metab Dis 2006;29:620–6.
29 Akiyama M, Titeux M, Sakai K et al. DNA-based prenatal diagnosis of harlequin ichthyosis and characterization of ABCA12 mutation consequences. J Invest Dermatol 2007;127:568–73.
30 McGrath JA, Gatalica B, Christiano AM et al. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2/COL17A1), a hemidesmosome transmembrane collagen, in generalized atrophic benign epidermolysis bullosa. Nature Genet 1995;11:83–6.
31 Pulkkinen L, Kimonis VE, Xu Y et al. Homozygous α6 integrin mutation in junctional epidermolysis bullosa with congenital duodenal atresia. Hum Mol Genet 1997;6:669–74.
32 Lane EB, Rugg EL, Navsaria H et al. A mutation in the conserved helix termination peptide of keratin 5 in hereditary skin blistering. Nature 1992;356:244–6.
33 Rothnagel JA, Dominey AM, Dempsey LD et al. Mutations in the rod domain of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 1992;257:1128–30.
34 Parmentier L, Clepet C, Boughdene-Stambouli O et al. Lamellar ichthyosis: further narrowing, physical and expression mapping of the chromosome 2 candidate locus. Eur J Hum Genet 1999;7:77–87.
35 Fischer J, Faure A, Bouadjar B et al. Two new loci for autosomal recessive ichthyosis on chromosomes 3p21 and 19p12-q12 and evidence for further genetic heterogeneity. Am J Hum Genet 2000;66:904–13.
36 Brambati B, Tului L. Chorionic villus sampling and amniocentesis. Curr Opin Obstet Gynecol 2005;17:197–201.
37 Evans MI, Andriole S. Chorionic villus sampling and amniocentesis in 2008. Curr Opin Obstet Gynecol 2008;20:164–8.
38 Fassihi H, Eady RA, Mellerio JE et al. Prenatal diagnosis for severe inherited skin disorders: 25 years’ experience. Br J Dermatol 2006;154:106–13.
39 Hohl D, Huber M, Frenk E. Analyses of the cornified cell envelope in lamellar ichthyosis. Arch Dermatol 1993;129:618–24.
40 Raghunath M, Hennies HC, Velten F et al. A novel in situ method for the detection of deficient transglutaminase activity in the skin. Arch Dermatol Res 1998;290:621–7.
41 Fine JD, Eady RAJ, Bauer EA et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB J Am Acad Dermatol 2008;58:931–50.
42 McGrath JA, Eady RAJ. The role of immunohistochemistry in the diagnosis of the non-lethal forms of junctional epidermolysis bullosa. J Dermatol Sci 1997;14:68–75.
Practical Aspects of DNA-Based Prenatal Diagnosis
The clinical application of DNA-based prenatal testing has had substantial benefits for families at risk for recurrence of further affected children with inherited skin disorders in subsequent pregnancies. Nevertheless, an important prerequisite to undertaking any DNA-based prenatal test procedure is the delineation of informative genetic markers. In most instances, it is important to have DNA samples from both parents and the affected individual to determine the pathogenic mutation(s) [1,2]. Availability of all these samples then permits screening for de novo
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


