AD, autosomal dominant; AR, autosomal recessive; BCC, basal cell carcinoma; SCC, squamous cell carcinoma, XR, X-linked recessive.
* Mutations in the SSH1 and SART3 genes have been reported in Chinese kindreds but not confirmed in subsequent studies.
Table 137.2 Hereditary skin disorders associated with mucocutaneous and extracutaneous malignancies. Muir–Torre syndrome, which can present with keratoacanthomas and sebaceous carcinomas in addition to internal malignancies, is included in Table 137.3 to allow juxtaposition with the constitutional mismatch repair deficiency syndrome. Diseases in bold text are covered in this chapter
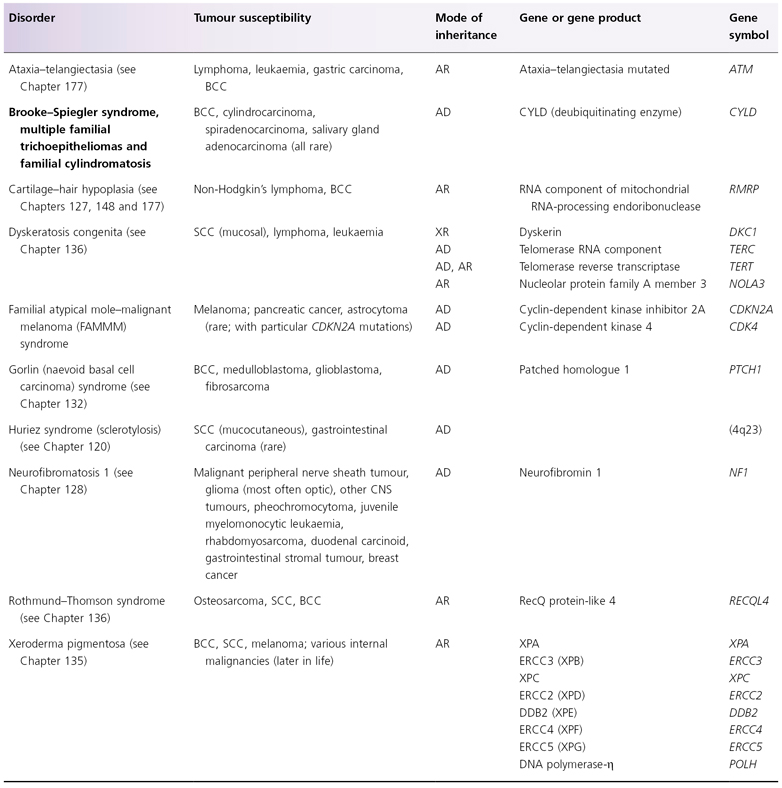
AD, autosomal dominant; AR, autosomal recessive; BCC, basal cell carcinoma; DDB2, DNA damage-binding protein 2; ERCC, excision repair cross-complementing; SCC, squamous cell carcinoma, XR, X-linked recessive.
Table 137.3 Hereditary skin disorders associated with extracutaneous malignancies. Diseases in bold text are covered in this chapter
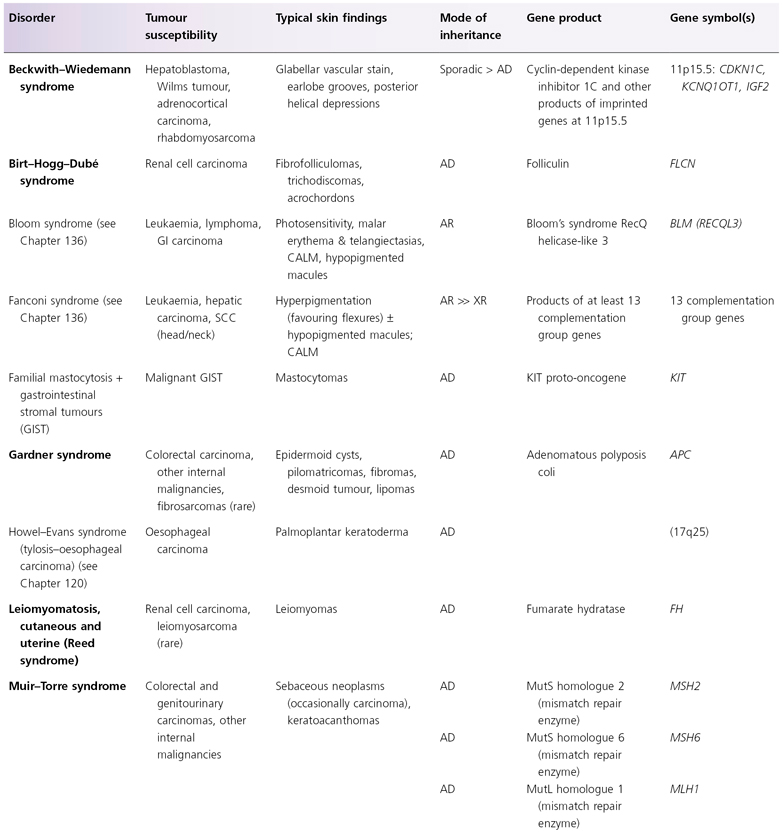
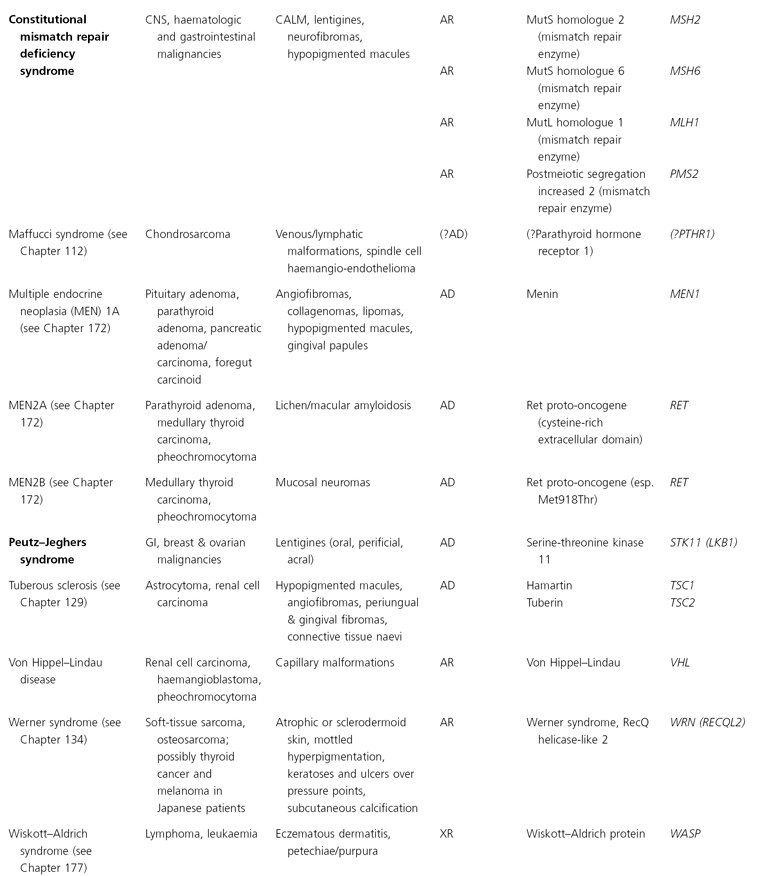
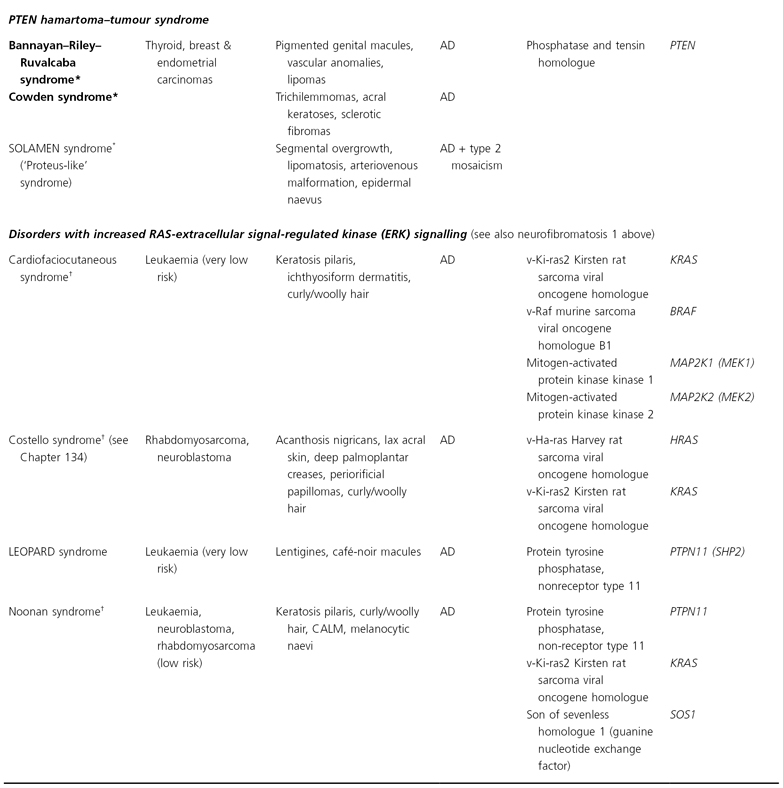
AD, autosomal dominant; AR, autosomal recessive; CALM, café-au-lait macules; CNS, central nervous system; GI, gastointestinal; LEOPARD, (syndrome of) lentigines, ECG changes, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, deafness; XR, X-linked recessive; SOLOMEN, (syndrome of) segmental overgrowth, lipomatosis, arteriovenous malformation, epidermal naevus.
* The findings of Bannayan–Riley–Ruvalcaba, Cowden and SOLOMEN syndromes overlap.
† Distal phalangeal transverse creases, especially on the volar thumbs, represent an additional feature.
Bazex–Dupré–Christol Syndrome
Definition.
The Bazex–Dupré–Christol syndrome (BDCS) is characterized by follicular atrophoderma, hypotrichosis, multiple milia, hypohidrosis, and early development of basal cell carcinomas (BCCs) [1,2].
Pathogenesis.
Bazex–Dupré–Christol syndrome is inherited in an X-linked semi-dominant manner, with more severe manifestations in male patients and milder findings in female patients. It has been mapped to chromosome Xq24–q27, but the causative gene has not yet been identified [3].
Clinical Features (Fig. 137.1).
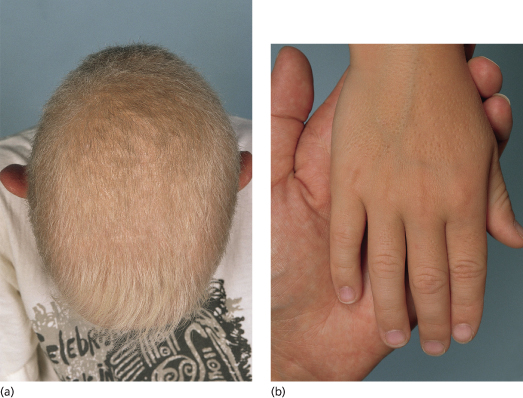
The congenital hypotrichosis of BDCS is diffuse in boys, while abnormal hairs are admixed with normal hairs in girls [4–6]. The eyebrows as well as scalp hairs may be affected, and hair shaft abnormalities can include trichorrhexis nodosa and twisted hairs mimicking pili torti. Numerous milia are often present in infancy and childhood. The milia have a predilection for the face but may also occur on the scalp, trunk and extremities. Follicular atrophoderma can be evident at birth or appear during childhood. It primarily affects the dorsal aspects of the hands and feet, the elbow and knees and (less often) the lower back and face. Exaggerated follicular or pseudo-follicular funnels (‘ice-pick’ marks) represent deep, wide ostia rather than true atrophy. Approximately half of BDCS patients have hypohidrosis, which is frequently limited to the head.
Fig. 137.1 Bazex–Dupré–Christol syndrome: (a) hypotrichosis and (b) follicular atrophoderma.
Courtesy of Professor Arnold Oranje.
Multiple BCCs usually begin to arise on the face during the second or third decade of life [4–6]. They often present as hyperpigmented papules that resemble melanocytic naevi but can exhibit locally aggressive behaviour. Basal cell naevi and multiple trichoepitheliomas on the face or in the genital area have also been described [7,8].
Differential Diagnosis.
Bazex–Dupré–Christol syndrome should be distinguished from the autosomal dominant naevoid BCC syndrome (Gorlin syndrome), which is due to defects in the PTCH gene and features palmoplantar pits, odontogenic keratocysts of the jaws, calcification of the falx cerebri, and skeletal abnormalities as well as early-onset BCCs (see Chapter 132). Rombo syndrome, like BDCS, manifests with hypotrichosis, milia, follicular atrophoderma and BCCs. However, Rombo syndrome can be differentiated by an autosomal dominant inheritance pattern, atrophoderma vermiculatum (i.e. follicular atrophoderma affecting primarily the face rather than the extremities), and cyanotic erythema of the face and hands.
The findings of BDCS also overlap with those of basaloid follicular hamartoma syndrome, an autosomal dominant condition that presents with hypotrichosis, milia, comedone-like lesions, palmoplantar pits, and skin-coloured to hyperpigmented papules (basal cell naevi) favouring the head and neck but not follicular atrophoderma or true BCCs. Of note, acrokeratosis paraneoplastica is also referred to as Bazex syndrome but represents a completely different disorder characterized by acral hyperkeratotic plaques in association with carcinomas of the upper aerodigestive tract.
References
1 Bazex A, Dupré A, Christol B. Genodermatose complexe de type indetermine associant une hypotrichose, un état atrophodermique généralisé et des degenerescences cutanées multiples (epitheliomas-basocellulaires). Bull Soc Fr Dermat Syph 1964;71:206.
2 Viksnins P, Berlin A. Follicular atrophoderma and basal cell carcinomas. The Bazex syndrome. Arch Dermatol 1977;113:948–51.
3 Vabres P, Lacombe D, Rabinowitz LG et al. The gene for Bazex–Dupré–Christol syndrome maps to chromosome Xq. J Invest Dermatol 1995;105:87–91.
4 Goeteyn M, Geerts ML, Kint A et al. The Bazex–Dupré–Christol syndrome. Arch Dermatol 1994;130:337–42.
5 Kidd A, Carson L, Gregory DW et al. A Scottish family with Bazex–Dupré–Christol syndrome: follicular atrophoderma, hypotrichosis and basal cell carcinoma. J Med Genet 1996;33:493–7.
6 Torrelo A, Sprecher E, Mediero IG et al. What syndrome is this? The Bazex–Dupré–Christol syndrome. Pediatr Dermatol 2006;23:286–90.
7 Yung A, Newton-Bishop JA. A case of Bazex–Dupré–Christol syndrome associated with multiple genital trichoepitheliomas. Br J Dermatol 2005;153:682–4.
8 Castori M, Castiglia D, Passarelli F et al. Bazex–Dupré–Christol syndrome: an ectodermal dysplasia with skin appendage neoplasms. Eur J Med Genet 2009;52:250–5.
Beckwith–Wiedemann Syndrome
Syn.
Exomphalos-macroglossia-gigantism syndrome
Definition.
Beckwith–Wiedemann syndrome (BWS) is characterized by visceral and somatic overgrowth, omphalocoeles and increased risk of early childhood malignancies [1,2].
Pathogenesis.
Beckwith–Wiedemann syndrome can result from a variety of genetic or epigenetic alterations that affect a cluster of imprinted growth regulatory genes on chromosome 11p15.5 [3,4]. Imprinted genes are normally expressed preferentially or exclusively from either the paternal or maternal allele. In some patients with BWS, loss of imprinting of the insulin-like growth factor 2 (IGF2) gene results in expression of both the maternal and paternal alleles of this growth-promoting gene instead of the normal expression of only the paternal allele [5]. Other changes at 11p15.5 that can lead to BWS include rare loss-of-function mutations in the cyclin-dependent kinase inhibitor 1C (CDKN1C) gene, more common loss of imprinting of the KCNQ1OT1 gene (resulting in expression of the maternal as well as paternal copy of an antisense RNA that downregulates CDKN1C), uniparental disomy that replaces the maternal alleles of all these genes with a second paternal copy, and rare chromosomal duplication (typically paternal) or translocation (typically maternal) [4,6,7]. BWS usually occurs sporadically, although autosomal dominant inheritance with preferential maternal transmission accounts for approximately 15% of cases [6,7].
Clinical Features.
The characteristic cutaneous manifestations of BWS are anterior earlobe grooves and circular depressions on the posterior helical rim [6,7]. A prominent glabellar vascular stain is another common finding. Extracutaneous features include macroglossia, midline umbilical wall defects (e.g. omphalocoele, umbilical hernia), organomegaly (e.g. liver, spleen, pancreas, kidneys, adrenal glands), and neonatal hypoglycaemia (due to increased insulin production). Somatic overgrowth can be prenatal and/or postnatal in nature and may result in gigantism or hemihypertrophy. Children with BWS have an increased risk of developing hepatoblastoma, Wilms tumour, rhabdomyosarcoma, adrenocortical carcinoma and neuroblastoma; overall, malignancy occurs in approximately 10% of patients [7,8].
Treatment.
Surveillance for malignancy includes serial abdominal ultrasounds and serum α-fetoprotein measurement during infancy and childhood [8].
References
1 Wiedemann HR. Complexe malformatif familial avec hernie ombilicale et macroglossie: un ‘syndrome nouveau’? J Genet Hum 1964;13:223–32.
2 Beckwith JB. Macroglossia, omphalocele, adrenal cytomegaly, gigantism, and hyperplastic visceromegaly. Birth Defects 1969;v:188–96.
3 Koufos A, Grundy P, Morgan K et al. Familial Wiedemann–Beckwith syndrome and a second Wilms’ tumor locus both map to 11p15.5. Am J Hum Genet 1989;44:711–19.
4 Weksberg R, Smith AC, Squire J et al. Beckwith–Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet 2003;12:R61–R68.
5 Weksberg R, Shen DR, Fei Y-L et al. Disruption of insulin-like growth factor 2 imprinting in Beckwith–Wiedemann syndrome. Nat Genet 1993;5:143–50.
6 Weksberg R, Shuman C, Smith AC. Beckwith–Wiedemann syndrome. Am J Med Genet 2005;137C:12–23.
7 Weksberg R, Shuman C, Beckwith JB. Beckwith–Wiedemann syndrome. Eur J Hum Genet 2010;18(1):8–14.
8 Zarate YA, Mena R, Martin et al. Experience with hemihyperplasia and Beckwith–Wiedemann syndrome surveillance protocol. Am J Med Genet 2009;149A:1691–7.
Birt–Hogg–Dubé Syndrome
Definition.
Birt–Hogg–Dubé syndrome (BHDS) is an autosomal dominant genodermatosis that features a classic triad of skin lesions – fibrofolliculomas, trichodiscomas and acrochordons – together with an increased risk of renal tumours and spontaneous pneumothoraces [1].
Pathogenesis.
Birt–Hogg–Dubé syndrome is caused by heterozygous loss-of-function mutations in the folliculin (FLCN; alias BHD) tumour suppressor gene [2]. More than 50 mutations in the FLCN gene have been identified, with almost all producing a truncated protein and approximately half involving insertion or deletion of a cytosine in a ‘hot spot’ hypermutable polycytosine tract in exon 11 [3–5]. Mutations in orthologous bhd genes underlie hereditary renal cancer in the Nihon rat and hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German shepherd dog. A high frequency of somatic inactivation of the germline wild-type FLCN/bhd allele has been noted in renal tumours from affected humans and rats.
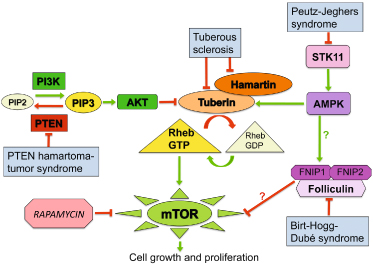
The folliculin protein is expressed in the kidney, lung and skin. Recently, novel folliculin-interacting proteins, FNIP1 and FNIP2, were identified [6,7]. FNIP1 binds to the AMP-activated protein kinase, a key energy sensing molecule that acts as a negative regulator of the mammalian target of rapamycin (mTOR) signalling pathway that stimulates cellular growth and survival (Fig. 137.2). The exact relationships between folliculin and mTOR activity remain to be determined [7].
Fig. 137.2 Mammalian target of rapamycin (mTOR) signalling in cancer-prone genodermatoses. The PTEN protein negatively regulates the PI3K/AKT pathway, which inhibits the GTPase activating-protein tuberin. Decreased tuberin activity leads to increased mTOR signalling and resultant cell growth. During periods of nutrient depletion (high AMP/low ATP), serine-threonine kinase 11 (STK11) promotes AMP-activated protein kinase (AMPK)-mediated activation of tuberin, which results in decreased mTOR signalling and less protein synthesis. Folliculin and folliculin-interacting protein 1 (FNIP1) form a complex with and are phosphorylated by AMPK, and mTOR activation (potentially via the AKT pathway) has been observed in renal tumors from Birt-Hogg-Dubé syndrome patients and (in some studies) folliculin-deficient mice. Increased mTOR signaling therefore contributes to the development of tumors in genodermatoses characterized by defects in PTEN, tuberin, STK11 or folliculin proteins. AMP, adenosine monophosphate; ATP, adenosine triphosphate; GDP, guanosine diphosphate; GTP, guanosine triphosphate; PIP2, phosphatidylinositol diphosphate; PIP3, phosphatidylinositol triphosphate; PTEN, phosphatase and tensin homologue deleted on chromosome 10.
Pathology.
In fibrofolliculomas, anastomosing strands of infundibular epithelium radiate from a distorted central follicle that is surrounded by a delicate fibromyxoid stroma. In trichodiscomas, the latter mesenchymal component predominates.
Clinical Features.
During the third or fourth decade of life, individuals with BHDS typically begin to develop multiple fibrofolliculomas, which affect >85% of patients, and (less often) trichodiscomas [5]. Both of these entities present as small, firm, whitish papules on the face, neck and upper trunk. Angiofibromas and perifollicular fibromas (folliculocentric lesions otherwise similar to angiofibromas) have been increasingly recognized in patients with BHDS and are thought to exist on a spectrum with fibrofolliculomas [8,9]. Soft, skin-coloured, acrochordon-like papules in flexural sites represent another finding in BHDS. Mucosal fibromas and additional mesenchymal skin lesions such as perivascular fibromas, collagenomas, focal cutaneous mucinosis and lipomas are also occasionally observed [3–5,8]. Cutaneous neoplasms that have been rarely reported in patients with BHDS include trichoblastoma, BCC, leiomyoma, leiomyosarcoma and dermatofibrosarcoma protuberans.
Individuals with BHDS have increased risk of developing renal tumours, particularly hybrid oncocytic and chromophobe renal carcinomas, which may be bilateral or multifocal. These tumours affect as many as a third of patients and develop at ages ranging from 20–70 years [3–5]. Spontaneous pneumothoraces are a frequent manifestation in BHDS; although most common in young adults, they have been reported in adolescents and children as young as 7 years [10]. Lung cysts are evident via computed tomographic scanning in approximately 85% of adults with BHDS [5]. Patients with BHDS may also be predisposed to the development of parotid gland tumours. Lastly, although colonic polyps and carcinomas were described as central features of the related and perhaps overlapping Hornstein–Knickerberg syndrome, which also manifests with perifollicular fibromas, a significantly increased incidence of colonic neoplasia has not been noted in large series of patients with BHDS [3–5].
Differential Diagnosis.
Birt–Hogg–Dubé syndrome shares several mucocutaneous features with tuberous sclerosis (which also involves dysregulation of the mTOR pathway; see Fig. 137.2) and multiple endocrine neoplasia type 1A, including facial angiofibromas, collagenomas and mucosal fibromas. Renal tumours and cystic lung disease represent additional overlapping manifestations of tuberous sclerosis and BHDS.
Treatment.
Recognition of the cutaneous manifestations of BHDS is important in order to facilitate early diagnosis of associated renal tumours and cystic lung disease.
References
1 Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 1977;113:1674–7.
2 Nickerson ML, Warren MB, Toro JR et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2002;2:157–64.
3 Schmidt LS, Nickerson ML, Warren MB et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 2005;76:1023–33.
4 Leter EM, Koopmans AK, Gille JJ et al. Birt-Hogg-Dubé syndrome: clinical and genetic studies of 20 families. J Invest Dermatol 2008;128:45–9.
5 Toro JR, Wei MH, Glenn GM et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 2008;45:321–31.
6 Baba M, Hong SB, Sharma N et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA 2006;103:15552–7.
7 Hartman TR, Nicolas E, Klein-Szanto A et al. The role of the Birt-Hogg-Dubé protein in mTOR activation and renal tumorigenesis. Oncogene 2009;28:1594–604.
8 Schaffer JV, Gohara MA, McNiff JM et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol 2005;53:S108–S111.
9 Misago N, Kimura T, Narisawa Y. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol 2009;36:943–51.
10 Bessis D, Giraud S, Richard S. A novel familial germline mutation in the initiator codon of the BHD gene in a patient with Birt-Hogg-Dubé syndrome. Br J Dermatol 2006;155:1067–9.
Brooke-Spiegler Syndrome, Multiple Familial Trichoepitheliomas and Familial Cylindromatosis
Definition.
Brooke–Spiegler syndrome (BSS), multiple familial trichoepitheliomas (MFT; epithelioma adenoides cysticum of Brooke) and familial cylindromatosis (FC) are overlapping, allelic autosomal dominant disorders characterized by a predisposition for the development of trichoepitheliomas, cylindromas and/or spiradenomas.
Pathogenesis.
Brooke–Spiegler syndrome, FMT and FC are caused by heterozygous loss-of-function mutations in the CYLD tumour suppressor gene [1–3]. These are typically truncating lesions or (less often) missense mutations affecting the ubiquitin-specific protease domain. There is no obvious genotype-phenotype correlation, with variable clinical findings and severity among kindreds with the same mutation and even within families [2,3]. The adnexal tumours that develop in affected individuals usually have somatic inactivation of the germline wild-type CYLD allele. The CYLD protein functions as a deubiquitinating enzyme (relatively specific for lysine 63-linked ubiquitin chains) and negatively regulates the nuclear factor-κB (NF-κB) and c-Jun N-terminal kinase (JNK) pathways that promote cell survival and proliferation [4].
Pathology.
Trichoepitheliomas, cylindromas and spiradenomas are tumours with follicular-apocrine differentiation [3]. Trichoepitheliomas feature small clusters or cribriform cords of basaloid cells with follicular germinative differentiation surrounded by dense fibrocytic stroma as well as small keratinizing cystic spaces with superficial follicular differentiation. Cylindromas, spiradenomas and spiradenocylindromas (hybrid tumours with features of both lesions) have apocrine lineage but are less obviously differentiated. Cylindromas display a ‘jigsaw puzzle’ pattern of angulated nests of basaloid cells enveloped by dense eosinophilic basement membrane material, while spiradenomas have larger, rounded nests composed of trabeculae with peripheral basaloid cells.
Clinical Features.
Brooke–Spiegler syndrome, MFT and FC exist on a spectrum and manifest between late childhood and early adulthood with progressive development of characteristic adnexal neoplasms [2,3]. Trichoepitheliomas and cylindromas occur together in BSS (sometimes along with spiradenomas), while trichoepitheliomas and cylindromas predominate in MFT and FC, respectively. Trichoepitheliomas are firm skin-coloured papulonodules with a predilection for the central face, especially the nose and melolabial folds (Fig. 137.3). Cylindromas present as slowly growing nodules that favour the scalp, referred to as ‘turban tumours’ in their fulminant form. Spiradenomas are less common and classically develop on the trunk and extremities as paroxysmally painful nodules with a slightly bluish hue. Syringomas have also been reported in individuals with BSS.
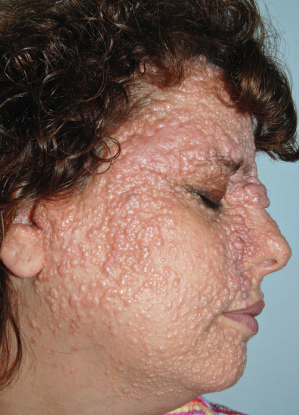
Cylindrocarcinomas and spiradenocarcinomas occasionally arise in patients with BSS-spectrum disorders and tend to behave aggressively, with destructive local growth and metastases [3,5]. BCCs have been noted to originate in continuity with trichoepitheliomas in BSS/MFT patients and adenocarcinomas (as well as adenomas) of the parotid and other salivary glands have also been observed [3].
Treatment.
Surgical excision, electrosurgical techniques and other ablative modalities (e.g. carbon dioxide or erbium:YAG laser treatment) can be used to control tumour burden [6,7]. Use of tumour necrosis factor-α inhibitors and aspirin to decrease the NF-κB signalling that is augmented in these disorders has also been described [7].
References
1 Bignell GR, Warren W, Seal S et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 2000;25:160–5.
2 Saggar S, Chernoff KA, Lodha S et al. CYLD mutations in familial skin appendage tumours. J Med Genet 2008;45:298–302.
3 Blake PW, Toro JR. Update of cylindromatosis gene (CYLD) mutations in Brooke-Spiegler syndrome: novel insights into the role of deubiquitination in cell signaling. Hum Mutat 2009;30:1025–36.
4 Courtois G. Tumor suppressor CYLD: negative regulation of NF-κB signaling and more. Cell Mol Life Sci 2008;65:1123–32.
5 Kazakov DV, Zelger B, Rutten A et al. Morphologic diversity of malignant neoplasms arising in preexisting spiradenoma, cylindroma, and spiradenocylindroma based on the study of 24 cases, sporadic or occurring in the setting of Brooke-Spiegler syndrome. Am J Surg Pathol 2009;33:705–19.
6 Rajan N, Trainer AH, Burn J et al. Familial cylindromatosis and Brooke-Spiegler syndrome: a review of current therapeutic approaches and the surgical challenges posed by two affected families. Dermatol Surg 2009;35:845–52.
7 Fisher GH, Geronemus RG. Treatment of multiple familial trichoepitheliomas with a combination of aspirin and a neutralizing antibody to tumor necrosis factor alpha: a case report and hypothesis of mechanism. Arch Dermatol 2006;142:782–3.
Epidermodysplasia Verruciformis
Definition.
Epidermodysplasia verruciformis (EDV) is an autosomal recessive condition characterized by genetic susceptibility to cutaneous infections with a particular group of human papillomavirus (HPV) types and the development of squamous cell carcinomas (SCCs) in affected skin.
Pathogenesis.
Patients with EDV are prone to infection with the β genus of HPV, which includes types 5, 8, 9, 12, 14, 15, 17, 19–25, 36–38, 46, 47, 49, 50, and others [1,2]. These HPV types are ubiquitous but generally do not cause clinical lesions in immunocompetent individuals. EDV patients are usually infected by multiple β-HPV types, and infections with other HPV types such as 3 and 10 can also occur. Particular HPV types may be associated with distinct skin lesion morphologies within an affected individual. HPV types 5 and 8 are most often detected in the SCCs that develop in EBV patients.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


