I Damage Recognition
Cellular DNA damage can be sensed and located in two ways [5] (see Fig. 135.1). The global genome repair (GGR) subpathway operates in a genome-wide manner but this is a slow DNA repair subpathway. Here, the XPC protein binds to the damage and initiates the further repair steps. The XPC protein is part of a heterotrimeric complex with hHR23B and centrin, and acts as the damage sensor. Another damage sensor in GGR is the UV-damaged DNA-binding protein (UV-DDB) consisting of the DDB1 and DDB2 subunits. DDB2 corresponds to the XPE gene product. DDB has a higher binding affinity and specificity for certain types of DNA damage than XPC and seems to assist XPC in detection of specific DNA damage such as cyclobutane pyrimidine dimers. Patients with XPC gene mutations usually develop a classic XP phenotype with skin cancer proneness but no neurological abnormalities. This may be due to the retained transcription coupled repair activity in XPC patients [4].
The stalled RNA polymerase II mediates damage recognition in actively transcribed genes. This second NER subpathway is called transcription coupled repair (TCR). TCR acts much faster than GGR (see Fig. 135.1). XPC and DDB proteins are dispensable in the TCR subpathway. Patients belonging to the XPC and XPE complementation groups therefore have normal TCR capabilities. In contrast, normal GGR but a deficiency in TCR are hallmarks of Cockayne’s syndrome (CS) patients (CSA or CSB gene defective, respectively). The exact functions of the CSA and CSB gene products still remain to be elucidated. CS proteins CSA and CSB may be involved in this recognition process but are not essential for transcription. CSB binds to DNA–RNA–RNA polymerase II complexes, and the resulting quaternary complexes are able to recruit transcription factor IIH (TFIIH), thus initiating the NER complex. The function of CSB may be to bring about the ubiquitin-mediated degradation of RNA polymerase II molecules stalled at lesions to enable repair to proceed. CSA has been shown to bind to the p44 subunit of TFIIH and CSB to bind to CSA, XPA, XPB, XPG and the p34 subunit of TFIIH. In general, the CS proteins may support the polymerase II complex to allow its temporary removal, and may play a general role in the processing of stalled polymerases during transcription. This general role in facilitating transcription may be one reason for the severe neurological abnormalities in some CS patients, whereas there are usually no neurological abnormalities in XPC or XPE patients [6].
II Damage Demarcation
The DNA double helix has to be unravelled around the damage to allow for excision repair. This is accomplished by the multifunctional basal TFIIH complex. TFIIH consists of 10 subunits including the XPB, XPD and TTD-A proteins. A 3′-5′helicase activity is a feature of the XPB protein and the XPD protein possesses a 5′-3′helicase activity. Trichothiodystrophy (TTD)-A was identified in 2004 and seems to play a role in regulating the level of TFIIH. If TTD-A is mutated, the autosomal recessive disease trichothiodystrophy results. This indicates that TTD results as a consequence of defective basal transcription, because TTD-A is not involved in NER. RNA polymerase II also utilizes TFIIH to initiate transcription at promoter sites. Thus, at least two different functions of TFIIH can be discerned, namely basal transcription and NER. The great phenotypic heterogeneity of defects in the XPD and XPB genes may be explained by this dual function of TFIIH (Fig. 135.2). Either NER or basal transcription or both may be disabled, depending on the type and location of the mutation within the respective gene. This can lead to clinical phenotypes that represent combinations of XP, TTD or even CS. The activity of TFIIH and stabilization of the ‘DNA bubble’ around the damage is supported by the replication protein A (RPA) (see Fig. 135.1). The XPA protein is recruited to the site of DNA damage later than TFIIH and complexes with RPA. XPA in conjunction with RPA may monitor DNA bending and unwinding, and verify the damage-specific localization of repair complexes rather than recognize DNA damage. However, the exact role of the XPA-RPA complex is not well understood. XPA patients usually are defective in GGR as well as in TCR which can explain the preponderance of XP with neurological symptoms, if XPA is mutated (see Fig. 135.2) [6].
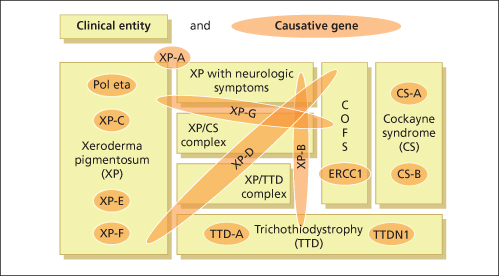
Fig. 135.2 Seven clinical entities based on 13 defective genes demonstrate the complex genotype-phenotype interactions.
Modified from Kraemer et al. 2008 [11].
III Incision of the Damaged DNA Strand
XPG and XPF (complexed with ERCC1), two endonucleases, are then recruited to incise the damaged DNA strand. The DNA strand at the 5′ boundary is cleaved by ERCC1-XPF and XPG cleaves at the 3′ boundary of the bubble structure (see Fig. 135.1). To allow precise positioning of the first DNA strand cut, XPG interacts with RPA and TFIIH. XPG is also required non-enzymatically for the subsequent 5′ incision by the XPF-ERCC1 heterodimer. As the lesion-containing DNA oligonucleotide that is excised is always from 24 to 32 nucleotides in size, this interaction between XPG and XPF may define the strand excision 24–32 nucleotides upstream. XPG is another multifunctional protein as it is also involved in base excision of oxidative DNA damage as a co-factor of the base excision protein OGGI. This may indicate why XPG patients often show neurological abnormalities, because oxidative DNA damage may persist in non-dividing neurones [7].
IV Removal of the DNA Damage-Containing Oligonucleotide
Always, an oligonucleotide of 24–32 nucleotides in size is excised by the sequential processes described above. And also always the DNA damage is located 5–6 nucleotides upstream of the 3′ end of the excised oligonucleotide. It is not known yet if the oligonucleotide is simply degraded after transport into the cytoplasm or if it subsequently induces UV-protective cell functions. Small oligonucleotides with specific sequences have been demonstrated to induce DNA repair as well as melanogenesis without UV exposure of the cells [8].
V Gap Filling
The excision process results in a single-strand gap which is filled by a newly synthesized oligonucleotide using the complementary strand as a template. This DNA repair synthesis involves DNA polymerase δ and/or polymerase ε activity, and depends on proliferating cell nuclear antigen (PCNA) and replication factor C (RFC). Reconstitution of in vitro repair synthesis was successfully accomplished using purified PCNA, RFC and either polymerase δ or polymerase ε. The very last step of NER consists of 3′ strand rejoining by DNA ligase I [1].
Normal repair of UV-damaged DNA requires the integrity of all the gene products mentioned above. Mutations in XPA–XPG and polymerase h (XPV) are responsible for the clinical phenotype of xeroderma pigmentosum. Mutations in CSA and CSB are responsible for the clinical phenotype of Cockayne’s syndrome. The clinical disease trichothiodystrophy results from mutations in TTD-A, XPD and XPB. Some non-photosensitive TTD patients have been identified who have a mutation in TTDN1, a gene of unknown function (see Fig. 135.2) [9].
References
1 Leibeling D, Laspe P, Emmert S. Nucleotide excision repair and cancer. J Mol Histol 2006;37:225–38.
2 Matsumura Y, Ananthaswamy HN. Toxic effects of ultraviolet radiation on the skin. Toxicol Appl Pharmacol 2004;195:298–308.
3 Kraemer KH, Ruenger TM. Genome stability, DNA repair and cancer. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ (eds) Fitzpatrick’s Dermatology in General Medicine. New York: McGraw-Hill, 2008: 977–86.
4 Thoms KM, Kuschal C, Emmert S. Lessons learned from DNA repair defective syndromes. Exp Dermatol 2007;16:532–44.
5 Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene 2002;21:8949–56.
6 Emmert S. Xeroderma pigmentosum, Cockayne syndrome, Trichothiodystrophy – defects in DNA repair and carcinogenesis. In: Allgayer H, Rehder H, Fulda S (eds) Hereditary Tumors. Weinheim: Wiley-VCH, 2009: 421–39.
7 Emmert S, Leibeling D, Runger TM. Syndromes with genetic instability: model diseases for (skin) cancerogenesis. JDDG 2006;4:721–33.
8 Eller MS, Asarch A, Gilchrest BA. Photoprotection in human skin – a multifaceted SOS response. Photochem Photobiol 2008;84:339–49.
9 Runger TM, DiGiovanna JJ, Kraemer KH. Hereditary diseases of genome instability and DNA repair. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ (eds) Fitzpatrick’s Dermatology in General Medicine. New York: McGraw-Hill, 2008: 1311–25.
Nucleotide Excision Repair Defective Syndromes
As discussed above, it is evident that cancer development is a multistep process. The process of tumour initiation is started if, for example, UV-induced DNA damage is not properly repaired. In terms of skin cancer development, the repair of UVB-induced pyrimidine dimers by the NER machinery prevails here. The clinical consequences of defective NER are vividly demonstrated by three major, but still rare, autosomal recessive inherited model diseases for UV-induced skin cancer: xeroderma pigmentosum (XP), Cockayne syndrome (CS) and the photosensitive form of trichothiodystrophy (TTD) (Table 135.1). Increased sun sensitivity and freckling in sun-exposed skin areas are clinical symptoms shared by all three clinical entities. However, XP patients differ from CS and TTD patients with respect to their proneness to skin cancer [1,2] (Table 135.2). The genotype-phenotype correlations are quite complex. In total, seven distinct clinical entities and 13 causative genes have been distinguished to date (see Fig. 135.2). Different mutations in one gene can give rise to more than one clinical entity (variable expression) and conversely, each clinical entity can arise from mutations in more than one gene (genetic heterogeneity).
Table 135.1 Nucleotide excision repair defective syndromes: clinical hallmarks and affected genes
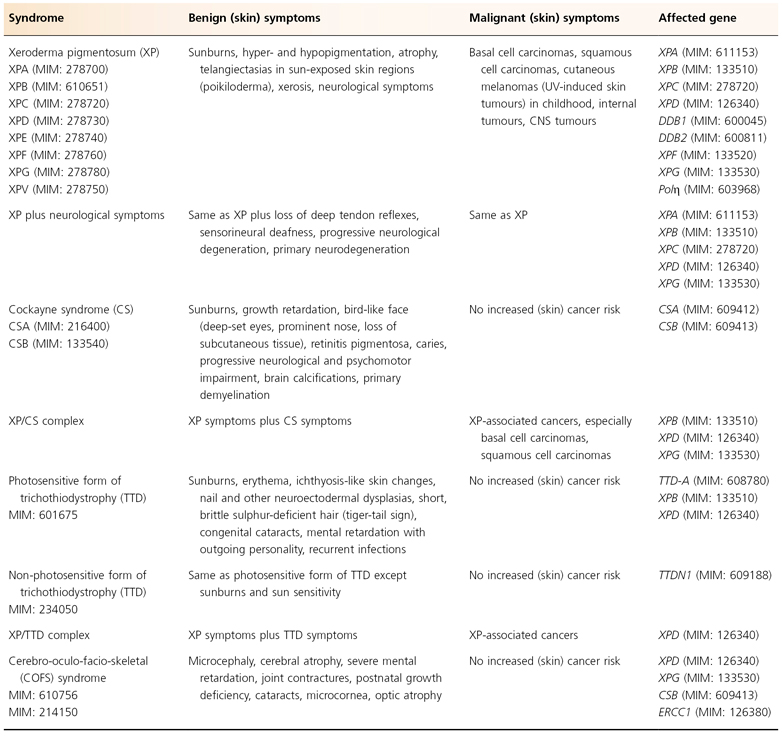
Table 135.2 Clinical features of nucleotide excision repair defective syndromes
Modified from Emmert (2009) [3].
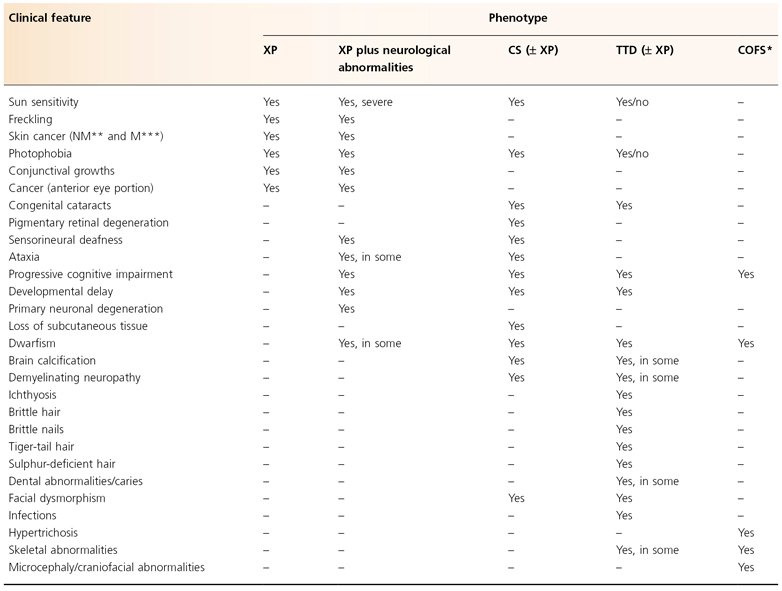
* Cerebro-oculo-facio-skeletal syndrome.
** Non-melanoma skin cancer (basal and squamous cell carcinoma).
*** Melanoma skin cancer.
Xeroderma Pigmentosum
History.
Moritz Kaposi described the first patient with ‘dry skin (xeroderma) and hyperpigmentations’ in 1863. This entity was finally named xeroderma pigmentosum in 1882 [4]. However, it was another 100 years before the pathogenic defect of XP was identified. Cleaver found in 1968 that cells from XP patients were defective in nucleotide excision repair. Normal cells repair UV-induced pyrimidine dimers by excising these dimers and replacing the gap in the DNA strand with newly synthesized DNA. DNA replication is normally restricted to the S phase of the cell cycle. Nucleotide excision repair, however, can occur at any time during the cell cycle. This phenomenon was called ‘unscheduled’ repair synthesis. The repair defect of XP cells was thus identified by showing that XP cells have decreased rates of unscheduled DNA synthesis. Eventually, it was shown by Epstein and colleagues in 1970 that all cell types in XP patients exhibit the excision repair defect including epidermal cells, fibroblasts, conjunctival cells, corneal cells and lymphocytes [1].
In 1972, de Weerd-Kastelein and colleagues fused together fibroblasts from different patients with XP to form heteropolykaryons (multinucleated cells containing nuclei from the two different strains). They were able to demonstrate correction of the excision repair defect in these heteropolykaryons, suggesting that different genetic defects could produce one similar phenotypic disease. Groups of patients whose cells failed to complement each other in culture were placed within a single complementation group. In this manner, seven complementation groups of XP have now been identified and are designated A–G. Again, it took more than two decades until the genetic basis of these complementation groups began to unravel. Today, the seven complementation groups, XPA to XPG, correspond to seven distinct XP genes, XPA–XPG [1,5].
Epidemiology.
The rare autosomal recessive inherited genetic disease XP occurs worldwide. Patients have been reported in all races, including whites, Asians, blacks and Native Americans. An examination of the literature demonstrated that 830 XP cases were reported from the first case in 1863 until 1982 [6]. In Japan, 91 cases were published, 210 cases in Europe, 49 cases in Egypt, and 241 cases in the USA. The incidence of XP in the USA and Europe is about 1:1 million [1,7]. Due to less mobile and more isolated populations, the incidence of XP is 10 times higher in northern Africa and Japan. Both sexes are affected equally. Consanguinity of the parents was present in about 30% of all cases [6].
If the patient carries homozygous, compound heterozygous or hemizygous mutations in one of the genes causing XP, clinical XP symptoms occur. Heterozygous carriers of XP mutations are regarded as ‘healthy’ to date. Notably, the frequency of such clinically normal individuals who are heterozygous carriers of a XP gene mutation is much higher (1:500) than XP patients (1:1,000,000). However, whether individuals heterozygous for a mutation in a XP gene are at increased risk of an UV-induced skin malignancy is not well understood. The only study to date of cancer risk in XP heterozygotes was published in 1979, before the XP genes were cloned [8]. This study was conducted on the pedigrees of XP families. The authors found that proposed carriers of one mutated XP allele have an elevated incidence of skin cancer.
Clinical Symptoms.
First XP symptoms (see Table 135.2) occur at a median age of 1–2 years [6]. Severe sun sensitivity is usually the first symptom that becomes apparent. However, approximately one-half of XP patients tan normally without excessive burning. In the other half, severe sunburn with blisters may develop after a very short sun exposure, even through window glass. The severe skin burning reaches the maximum after 24 hours and then resolves within the next 4–6 days, lasting longer than in normal healthy individuals. Infants may be misdiagnosed as being scalded by doctors who then initiate investigations concerning child abuse. In later stages of infancy, pigmentary changes in sun-exposed skin develop. Poikiloderma becomes apparent, including telangiectasias and atrophic hyper- as well as hypopigmentations (Fig. 135.3). This is a sign of premature skin ageing, because poikiloderma normally develops after chronic sun exposure over decades. The poikilodermic skin changes are usually sharply demarcated from sun-protected skin.
Fig. 135.3 Clinical symptoms of XP. (a) Skin changes sharply demarcated to sun-exposed skin. (b) Typical poikilodermic aspect of XP skin changes including the lips (atrophic dry skin with hyper- and hypopigmentations and telangiectasias). (c) Involvement of the anterior eyes (pterygium). (d) Child with classic XP symptoms and large squamous cell carcinoma on the left cheek. (e) Melanoma on XP skin (arrow). (f) Basal cell carcinoma on XP skin (arrow).
Reproduced from Emmert [3] with permission from Wiley-Blackwell.
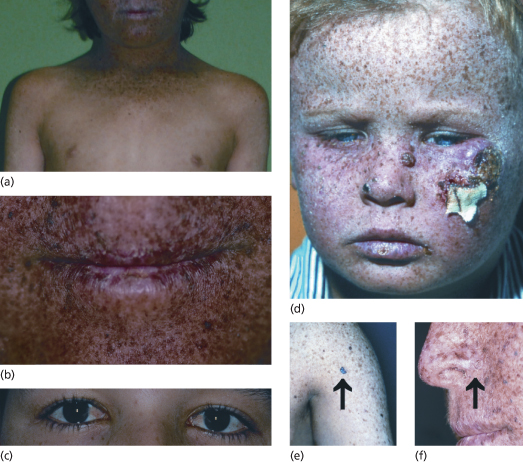
Later on, in early childhood, the first skin cancers appear that include all common UV-induced skin cancers such as basal and squamous cell carcinoma and cutaneous melanoma (see Fig. 135.3). Basal cell carcinomas and squamous cell carcinomas of the face, head and neck account for 80% of all cutaneous malignancies in the USA. In patients with XP, 97% arise on these sun-exposed skin sites. The risk of developing a cutaneous malignancy, including melanoma, under 20 years of age is increased 1000-fold in XP patients. In a retrospective study of more than 830 XP patients, the median age of first skin cancer development was assessed as 8 years. In contrast, the median age of first skin cancer development in the normal Caucasian population is about 60 years [1,9]. There is no method or marker to predict the severity of the disease. The age of onset of symptoms is no marker for disease progression, nor is the progression predictable within family members [10].
Ocular Manifestations
Xeroderma pigmentosum patients also suffer from UV damage to the sun-exposed portions of the eyes, as the repair defect is present in all cells of the body. Photophobia, conjunctivitis, keratitis, pterygium (see Fig. 135.3) and neoplasias of the eyelids and conjunctivae may develop. The posterior portions of the eye, such as the retina, are usually unaffected because only visible light (400–800 nm) reaches the retina and UV light is absorbed by anterior portions of the eye. Therefore, XP patients may want to wear UV-protective glasses with side shields. The cornea can be moisturized with methyl cellulose eyedrops. Patients with corneal opacity from severe keratitis may benefit from corneal transplantations but there is a risk of graft rejection due to neovascularization. In addition, atrophic rash, telangiectasias and neoplasias at the tip of the tongue reflect relevant sun exposure to that area [1].
Neurological Symptoms
Xeroderma pigmentosum patients may additionally develop neurological symptoms in about 20% of cases (see Fig. 135.2). There is great variability in the onset and severity of these neurological symptoms, but all share a progressive character (see Table 135.2). The onset may be in early infancy or delayed until puberty and the neurological abnormalities may be mild or severe. Reduction or loss of deep tendon reflexes, followed by progressive deafness, are normally the first neurological symptoms. Then, mental deterioration with disabilities in speaking, walking and balance may follow.
Neurological deficiency in XP was first reported by DeSanctis & Cacchione in 1932. The patients described were three severely affected Italian brothers with intellectual impairment, microcephaly, delayed motor development, sensorineural deafness, peripheral neuropathy and, finally, dementia, beginning at 2 years of age. Patients with DeSanctis–Cacchione syndrome have XP and severe neurological progressive deficiencies, including choreo-athetosis, Achilles tendon shortening leading to eventual quadriparesis, and immature sexual development. These patients represent a subgroup of complementation group A. In clinical practice, deep tendon reflexes testing and routine audiometry usually can serve as a screen for the presence of XP-associated neurological abnormalities. In cases where there is clinical evidence of early neurological abnormalities, a brain magnetic resonance imaging (MRI) scan may show enlarged ventricles. The pathological correlate of the neurological symptoms is a primary axonal degeneration with loss of neurones, particularly in the cerebrum and cerebellum (see Table 135.2). XP patients with defects in the XPA, XPB, XPD or XPG gene are mostly affected with additional neurological abnormalities but the molecular basis for these is still under investigation. One possible explanation would be that they are due to an accumulation of endogenous, e.g. oxidative, DNA damage to which non-proliferating neurones would be most susceptible. It was found that some XP genes are also involved in BER of oxidative DNA damage. In parallel, there are indications that some oxidative DNA damage is repaired via the NER system [3,11].
Internal Neoplasias
The risk of development of internal neoplasias may also be increased in XP patients (see Table 135.1). It has been estimated that the overall risk of non-cutaneous malignancy is increased approximately 10–20-fold. Primary brain tumours, sarcomas, leukaemia and lung cancers have been reported in XP patients [3.] Benzapyrene derivates induce DNA damage that is repaired by NER. Thus, tobacco smoke may be regarded as ‘internal sun’ for XP patients [1]. It is recommended that XP patients refrain from smoking and that parents should protect children with XP from being exposed to second-hand smoke. XP may also be associated with metabolic diseases. In 1998, an XP patient with a splice site mutation within the XPC gene was reported, who suffered from hypoglycinaemia [12]. This may comprise a new syndrome analogous to the XP/CS complex syndrome (see below).
The development of skin cancer, progressive neurological symptoms and resulting complications drastically reduces the life expectancy of XP patients, who die 30 years earlier than healthy individuals on average. It has been estimated that the probability of a 40-year life expectancy for XP patients is about 70% [6].
Histopathology.
The histological features of XP patients are similar to the features of severe sun damage. Hyperkeratosis, atrophy of the epidermis and irregular elongation or atrophy of the rete ridges can be found. Irregular hyperpigmentation of the basal layer, either with or without an increase in the number of melanocytes, is often present. Hypopigmented macules in between show decreased numbers of melanocytes. A non-specific chronic inflammatory infiltrate may be present in the upper dermis. In addition, typical histologies of UV-induced malignant skin tumours may be seen, including cutaneous melanoma, basal cell and squamous cell carcinoma, and keratoacanthoma. Taken together, the histological features resemble sun-damaged aged skin but are non-specific for XP.
Differential Diagnoses.
The diagnosis of XP should be established as early as possible to implement UV-protective measures. Photosensitivity, often expressed as crying on exposure to sunlight in an infant, and sunburn comprise early signs. Such symptoms may also be found in differential diagnoses such as erythropoietic protoporphyria, erythropoietic porphyria, polymorphous light eruption, Rothmund–Thomson syndrome and Hartnup or Bloom syndrome (Box 135.1). Drug-induced photosensitivity or severe sunburn may also result in such symptoms. Biopsies and examination of the urine, stool and blood for porphyrins should be performed to help with clarification of these differential diagnoses. As pigmentation is noted, other differential diseases may come to mind. These include urticaria pigmentosa, dyskeratosis congenita, radiodermatitis, Carney complex or other premature ageing syndromes such as progeria. The occurrence of malignancies may resemble basal cell naevus syndrome or familial atypical multiple mole–melanoma (FAMMM) syndrome (see Box 135.1).
Box 135.1 Potential differential diagnoses of nucleotide excision repair defective syndromes
Xeroderma Pigmentosum
- Numerous ephelids on untypical localizations, e.g. lips
- Polymorphous light eruption
- Drug-induced photosensitivity
- Hydroa vacciniforme
- Subacute cutaneous lupus erythematosus
- Erythropoietic porphyria
- Urticaria pigmentosa
- Basal cell naevus syndrome (Gorlin–Goltz syndrome)
- FAMMM syndrome
- Dyskeratosis congenita
- LEOPARD syndrome
- Hartnup disease
- Progeria (Werner syndrome)
- Rothmund–Thomson syndrome
- Bloom syndrome (congenital telangiectatic erythema)
- Carney complex
Cockayne Syndrome
- Chromosomal abnormalities
- Hallermann–Streiff syndrome
- Russell–Silver syndrome
- Brachmann–deLange syndrome
- Dubowitz syndrome
- Tyrosinaemia
- Leucodystrophies
- Peroxisomal disorders
Trichothiodystrophy
- Ectodermal dysplasias
- Hidrotic (Clouston) ectodermal dysplasia
- Ichthyoses
Laboratory Tests.
In addition to the clinical hallmarks of sun sensitivity, freckling and proneness to skin cancer, XP may be diagnosed by further laboratory tests [13]. These include functional assessment of cellular repair capacity and genetic tests (mutational analysis). Cell lines from XP patients can be established either from blood (virus-transformed lymphoblastoid cell lines) or from a skin punch biopsy (primary fibroblast cell lines). Such cultured cells grow normally when not exposed to damaging agents. However, after UV irradiation, the population growth or the colony-forming ability is reduced to a greater extent than normal. Cells from XPV patients show this phenomenon only in the presence of caffeine in the culture medium which is a hallmark for XPV cells. The cellular NER capacity can be measured with a host cell reactivation assay. Here, UV-damaged viruses or DNA plasmids are brought into the patient cells. In every form of XP cell tested, there is a deficiency in the ability to repair the virus or plasmid to a functionally active state compared to normal cells. Such functional tests may be followed by genetic investigations on the base sequence, mRNA and protein level. In general, XP cells have a normal karyotype without excessive chromosome breakage or increased sister chromatid exchange. If a mutation is known in an affected family, prenatal diagnosis by amniocentesis in future pregnancies is possible using cells from amniotic fluid.
Treatment.
The treatment of XP patients consists of three main steps: making the diagnosis as early as possible, rigorous sun protection, and regular skin examination with treatment of all premalignant and malignant skin changes. Sun protection includes very strict sun avoidance and wearing protective clothing, UV-absorbing glasses and long hairstyle. Special suits have been developed that completely block UV but consist of light fabrics. Patients should adopt a lifestyle to minimize UV exposure and use sunscreens with high sun protection factor (SPF) ratings daily (best SPF 50+). Some families use UV meters to measure the UV content in their environment. Such measures should be paralleled by careful and frequent skin examinations for early skin cancer detection. Ideally, a family member should be instructed in recognition of cutaneous neoplasms. A complete (digital) photodocumentation of the entire skin surface with additional close-ups (e.g. by digital epiluminescence microscopy) of lesions is often useful in detecting new lesions. A dermatologist should examine patients at short intervals (every 3–6 months).
The most common premalignant skin lesions are actinic keratoses. These can be treated with the common treatment modalities listed in treatment guidelines, including cryotherapy, curettage or topical 5-fluorouracil. For the treatment of larger skin areas, topical use of imiquimod is also feasible. Caution is recommended, however, in the use of photodynamic therapy or other light- or laser-based therapies, because an abnormal response cannot be excluded in XP patients. Therapeutic dermabrasion or dermatome shaving has been applied to remove the more UV-damaged superficial skin layers and to allow repopulation of these layers by more UV-shielded cells from hair follicles or glands. That this is a valid hypothesis is demonstrated by patients who received a larger skin graft to the face. The graft taken from sun-protected areas like the buttocks or upper thighs is often resistant to the development of skin cancers for many years compared to the surrounding ungrafted skin.
If skin cancer occurs, it should be treated according to guidelines, with surgical excision being first choice as it is for patients who do not have XP. As XP patients may need hundreds of surgical excisions on their face over a lifetime, removal of undamaged skin should be minimized. This may be accomplished by Mohs’ micrographic surgery. Interestingly, most XP patients are not abnormally sensitive to therapeutic X-rays and respond normally to full doses of therapeutic X-radiation for treatment of inoperable neoplasms. However, because some XP patient cells show hypersensitivity to X-rays, it may be prudent to test for clinical X-ray hypersensitivity with an initial low dose. XP patients should not smoke or be exposed to second-hand smoke, as tobacco smoke produces DNA lesions that are repaired by the NER system.
High-dose oral isotretinoin can significantly reduce the frequency of skin cancers at least in some XP patients. Due to its side-effects (hepatic, hyperlipidaemic, teratogenic, calcification of ligaments and tendons, premature closure of the epiphyses), this treatment may be reserved for XP patients who are actively developing large numbers of new skin cancers. However, after cessation of isotretinoin the skin cancer frequency may recur. The topical use of T4 endonuclease V, a prokaryotic DNA repair enzyme that initiates repair of UV-induced CPDs, has been shown to reduce the rate of skin cancers (actinic keratoses and basal cell carcinomas) in patients with XP. To deliver the agent, T4 endonuclease V is encapsulated in a liposomal lotion. This treatment on a daily basis is well tolerated and no side-effects have been reported so far. However, it is still under research and has not been approved for clinical use [5,14].
Complementation Groups.
Two clinical entities can be distinguished in patients with XP symptoms: XP and XP with neurological abnormalities (see Fig. 135.2). Besides classification according to the presented clinical phenotype, XP patients can also be classified according to their mutated gene. Seven XP complementation groups, XPA to XPG, can be discerned according to this genotypic classification (Table 135.3). XP patients with neurological symptoms typically fall into complementation groups XPA, XPB, XPD or XPG. Between groups as well as within a single group, there can be great variability in the severity of symptoms [1,15]. This variability depends on the special type and location of the respective XP gene mutation and the extent to which one or more XP gene functions are disrupted. These complex genotype-phenotype relations and the roles of DNA repair genes in transcription and other functions are currently being investigated [16].
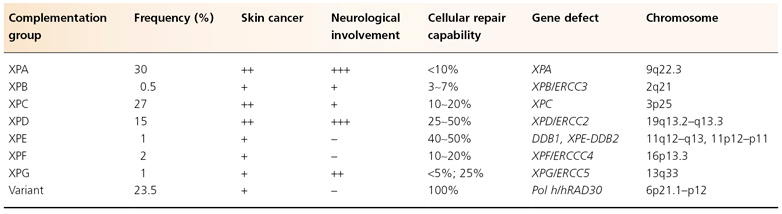
Table 135.3 Characteristics of the seven XP complementation groups and XP variant
Complementation Group A
Xeroderma pigmentosum patients in this group represent the second largest group worldwide. Patients are reported from Japan, the USA, Europe and the Middle East. Often, severe XP symptoms are seen in this group with an early age of onset. Additional neurological symptoms are common, although subsets of patients within this group have been reported to develop minimal or no neurological abnormalities at all. The most severely affected patients, those with the DeSanctis–Cacchione syndrome, are represented in this group. XPA patients are totally deficient in NER.
The XPA gene has been identified on chromosome 9q22. It contains 273 amino acids and binds specifically to damaged DNA via a zinc-binding domain in the C-terminal region. Although this protein was originally hypothesized to be responsible for the recognition of damaged DNA, recent studies have demonstrated that it is rather involved in damage verification and recruitment of other NER subunits such as RPA and TFIIH (see Fig. 135.1). Many mutations have been identified within the XPA
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


