Fig. 132.2 The HH–PTCH–SMO pathway (a postulated view incorporating information from Drosophila, mouse and human). (a) In cells not exposed to HH, PTCH blocks SMO from entering the primary cilium and becoming activated. The activation of Gli (part of a tetrameric complex at the microtubules) is inhibited. (b) In the presence of hedgehog, the inhibition of SMO by PTCH is released and SMO migrates from intracellular endosomes to the cell membrane of the cilium. SMO is activated in the cilium and activates a signal transduction cascade resulting in mature active Gli being released from the tetrameric complex to promote transcription of target genes. The patched/hedgehog complex is internalized and degraded or destabilized.
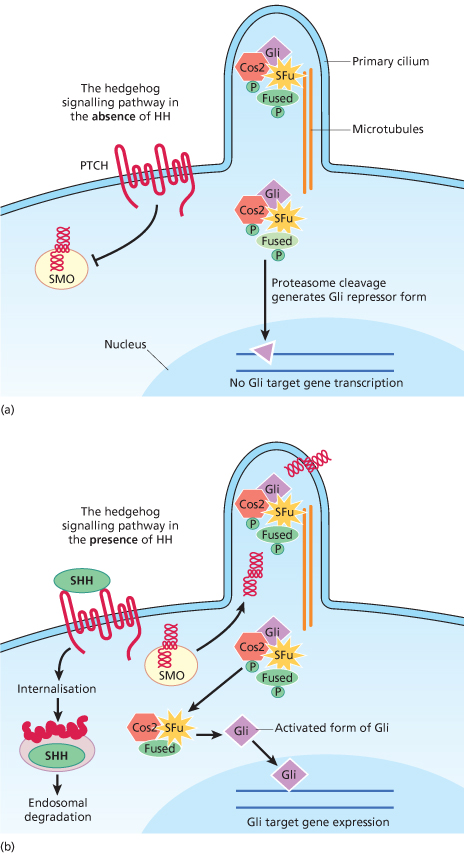
When extracellular SHH binds to PTCH, the inhibition of SMO is released, activating the signalling pathway and transcription of downstream target genes (Fig. 132.2b).
The SHH–PTCH–GLI pathway appears to be sensitive to the levels of its various proteins. Any mutation or polymorphism in one or more of the genes may affect the amount of functional protein, so affecting the activation or repression of downstream genes. A wide range of variation in transcription could therefore result from different levels of activity of PTCH. There is experimental evidence for this: complete inactivation of one allele (resulting in 50% patched activity) leads to the features of Gorlin syndrome in mice [27] and PTCH missense alleles produce active proteins but with reduced activities [28]. That the pathway is dosage sensitive may explain the spectrum of clinical presentation, as seen between and within families in Gorlin syndrome.
The jaw cysts and basal cell carcinomas in Gorlin syndrome are associated, however, with a different mechanism – loss of function of the usual PTCH allele [29,30], thereby releasing the cell from the remaining control of the SHH–PTCH–GLI pathway exerted by that allele. In fact, inactivation of PTCH or oncogenic activation of SMO occurs in almost all non-syndromic BCCs as well, suggesting that dysregulation of SHH signalling is a prerequisite for BCC formation [25].
A wide spectrum of PTCH mutations has been found in patients with Gorlin syndrome [31]. The mutations are spread throughout the whole coding region, with no apparent clustering.
Mutations are detected in about 85% of patients who meet the diagnostic criteria. The detection rate is lowest in people who are the first affected individual in their family, most probably because of somatic mosaicism. The mutation may often be more easily detected if an affected child is tested. For patients in whom there is a clinical suspicion of mosaicism, detecting the same PTCH mutation in several tumours but not in lymphocyte DNA may confirm this.
The frequency of classes of mutations, obtained from the literature [21,22,32–35] and the DNA Diagnostic Laboratory at Birmingham Women’s Hospital, UK, is 65% truncating, 16% missense, 13% splice site mutations and 6% intragenic or large-scale deletions or rearrangements.
There appears to be no genotype–phenotype correlation with truncating mutations [35]. It is not possible to make predictions about clinical severity for developmental and neoplastic features associated with specific mutations because of the likely modifying effects of other genes and environmental factors.
PTCH germline mutations have not been associated with any other heritable syndromes, but somatic mutations have been found in a range of sporadically occurring tumours including those observed in Gorlin syndrome: non-syndromic BCC, skin trichoepithelioma, medulloblastoma, ovarian fibroma and keratocysts.
Missense mutations of PTCH have been reported in 5% of unrelated probands with holoprosencephaly [36]. The authors hypothesized that the missense mutations would lead to enhanced PTCH repressive activity on the hedgehog signalling pathway, unlike the mechanism in NBCC in which the pathway is activated.
Pathology
Histology of Basal Cell Carcinomas
The naevi and NBCCs are histologically identical to BCCs. About one-third of patients have two or more types of NBCCs, including superficial, multicentric, solid, cystic, adenoid and lattice-like [37]. NBCCs are more commonly associated with melanin pigmentation and foci of calcification than non-syndromic BCCs, but otherwise cannot be distinguished.
Histology of Jaw Cysts
The histological features are characteristic [38]. The cysts are lined by a parakeratotic stratified squamous epithelium, which is usually about 5–8 cell layers thick and without rete ridges. Rarely, the form of keratinization is orthokeratotic. The basal layer is well defined with regularly orientated palisaded cells. Satellite cysts, epithelial rests and proliferating dental lamina are sometimes seen in the cyst capsules.
Palmar and Plantar Pits
The pits appear to be caused by premature desquamation of horny cells along the intercellular spaces, but are not due to degeneration of the horny cells themselves. Light microscopy reveals a lack of keratinization of pit tissue and a proliferation of basaloid cells in irregular rete ridges [39].
Electron microscopy [40] showed that the epithelium at the base of the pits consists of keratinocytes containing poorly developed tonofibrils. Discharge of cementsomes from the horny cells was incomplete, perhaps due to a shortened transit time of keratinocytes, so failing to provide enough intercellular cement.
Scanning electron microscopy [39] showed the stratum corneum overlying the epithelium at the bases of the pits to be thin, irregular and containing large defects. The stratum corneum at the margins of the pits was thicker, more compact and more adherent. The pit walls descended from a gently rounded top to a point of sharp transition between pit epithelium and normal epithelium.
Response to Radiation.
As some patients respond to therapeutic irradiation by later developing crops of BCCs in the treated area, in vitro studies of cellular radiation hypersensitivity have been undertaken, with conflicting results. It appears, however, that the cancer susceptibility is neither caused by nor manifested as chromosome instability, or that increased cell killing is a major effect of the gene.
Little et al. [41] demonstrated a moderate degree of radiation hypersensitivity in one family member whereas the remaining affected and non-affected individuals from the same family responded normally. They suggested that isolated cases of in vitro radiation hypersensitivity might not relate to the underlying genetic disorder. It seems likely that the response to radiation may be affected by other genes of major effect, and their delineation by comparing family members with the syndrome would be useful research.
There is supporting evidence in an animal model for the adverse effects of radiation. With age, mice heterozygous for an inactivating PTCH mutation spontaneously developed BCC-like tumours [42]. However, the BCCs were of far greater number and size in mice that had received ultraviolet irradiation. A single dose of ionizing radiation markedly enhanced development of BCCs.
Ultraviolet Radiation
Circumstantial evidence that exposure to sunlight may be deleterious comes from population studies: 14% of cases in a north-west England study [10] developed a BCC before the age of 20 years, compared with 47% in Australia (G. Trench, personal communication).
Laboratory experiments with ultraviolet (UV) radiation, however, have been inconclusive. Fibroblasts have been found to have no differences in sensitivity [43] following ultraviolet C (UVC), whereas others were more sensitive to UVC [44].
The majority of experiments have been conducted with UVC radiation (254 nm), but epidemiological and clinical studies indicate that UVB radiation (280–320 nm) in sunlight is responsible for the induction of most skin cancers in humans.
Gorlin syndrome fibroblasts have been shown to be hypersensitive to killing by UVB but not UVC radiation [45,46] compared with skin fibroblasts from normal individuals. This was not due to a defect in the excision repair of pyrimidine dimers [46].
Clinical Features and Natural History.
The major features of the syndrome which present in childhood are shown in Table 132.1, compiled from a review of the literature and the author’s own observations of over 200 patients, many of whom have been followed for more than 10 years. Some features are described in more detail below. Several large studies all give similar results [10,12,47,48].
Table 132.1 Frequency of features in childhood Gorlin syndrome by organ system
| Features | % |
| Skin | |
| Milia | 42 |
| Meibomian cysts | 6 |
| Epidermal cysts | 44 |
| Skin tags | 6 |
| Palmar pits | |
| <10 years | 65 |
| <15 years | 80 |
| Plantar pits | 49 |
| Naevi <20 years | 53 |
| Basal cell carcinomas | |
| <20 years | 14 |
| >20 years | 73 |
| >40 years | 92 |
| Jaw cysts | |
| 10 years | 13 |
| 20 years | 51 |
| >40 years | 79–90 |
| Mis-shapen/missing teeth | 30 |
| Skeletal | |
| High arched palate | 6 |
| Sloping shoulders | 61 |
| Sprengel shoulder | 46 |
| Pectus excavatum | 20 |
| Thoracic scoliosis | 47 |
| Short fourth metacarpal | 26 |
| Short terminal phalanx thumbs | 9 |
| Stiff thumbs | 6 |
| Polydactyly | 8 |
| Occipitofrontal head circumference | 97 |
| Operative delivery | 62 |
| Facial features | |
| Bossing | 79 |
| ‘Typical face’ | 70 |
| Prognathism | 46 |
| Eyebrows arched | 28 |
| Palpebral fissures downslanting | 30 |
| Upslanting | 13 |
| Eye anomalies | 30 |
| Strabismus | 19 |
| Cataracts | 4 |
| Cleft lip and palate | 7 |
| Other features | |
| Mental retardation | ?3 |
| Epilepsy | 6 |
| Medulloblastoma | 5 |
| Ovarian fibroma | 24 women |
| Undescended testes | 6 males |
| Inguinal hernia | 17 males |
| Cardiac fibroma | 2.5 |
| X-ray findings | |
| Cervical/thoracic vertebral anomalies | 60 |
| Rib anomalies | 70 |
| Calcification: falx <15 years | 40 |
| Diaphragm sella (20 years) | 100 |
Developmental History
Of children in the author’s series, 62% had an operative delivery. The average birthweight was 4.1 kg and head circumference was 38 cm, both greatly increased when compared with siblings. Walking was delayed until an average of 18 months; siblings walked at 12–13 months. Several children had investigations for hydrocephalus because of striking macrocephaly – the head circumference was grossly above the 97th centile but growth continued parallel with the centile lines. Many children initially have a motor delay, but appear to catch up. All children known to the author have attended mainstream school, a few needing additional help.
In the literature, ‘mental retardation’ has been reported in about 3%. In the population study in the north-west of England, there were no cases of moderate or severe mental retardation in 84 cases [10], apart from treated cases of medulloblastoma. About 6% of patients in that study required prolonged anticonvulsant therapy for grand mal seizures.
Build
Patients tend to be tall. Their heights are usually above the 97th centile, often in marked contrast with unaffected siblings. Some patients exhibit a marfanoid build.
Shape and Size of Cranium
One of the most striking features is the increased head size present from birth. All children and adults in the author’s series had a head circumference on or above the 97th centile and above the corresponding centile line for height. The head gives the appearance of being long in the anteroposterior (AP) plane, with a prominent and low occiput.
Facial Appearance
About 70% of patients have the characteristic facies but there is intrafamilial variation. Figures 132.3 and 132.4 show children and an adult with the syndrome. Frontal, temporal and biparietal bossing in 80% give a prominent appearance to the upper part of the face, and patients often adopt hairstyles that disguise the bossing. There is often facial asymmetry. Some patients have well-developed supraorbital ridges, giving the eyes a sunken appearance. The eyebrows are often heavy, fused and arched. There is a broad nasal root and hypertelorism. The inner canthal, interpupillary and outer canthal distances are all generally above the 97th centile, but appear to be in proportion with the head circumference. The mandible is long and often prominent with the lower lip protruding in front of the upper. There is a well-established association with cleft lip/palate, which occurs in 5–6%.
Fig. 132.3 Four children with Gorlin syndrome showing the degree of variation in facial appearance, frontal bossing and sloping of the shoulders. Sprengel shoulder is also shown (b).
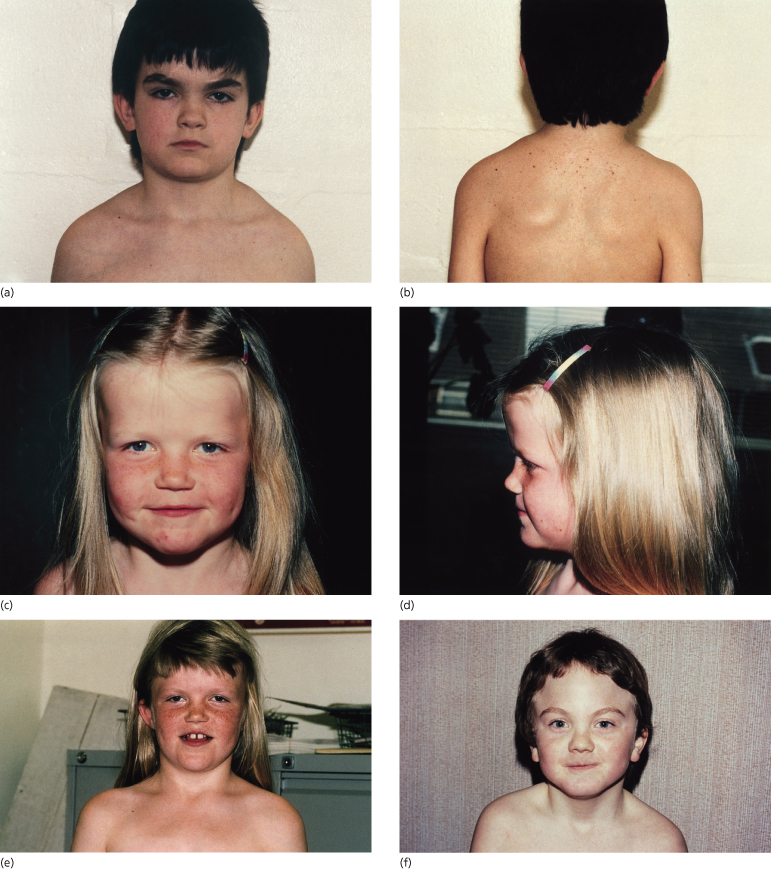
Fig. 132.4 An adult patient with Gorlin syndrome showing many of the facial features associated with the syndrome – arched eyebrows, downslanting palpebral fissures and sloping shoulders. Frontal bossing is disguised by the cut of the hair.

It can be very helpful to compare a person’s facial appearance with siblings because there is usually a striking difference in the facial gestalt between unaffected and affected siblings.
In a personally examined series, 26% of cases had ophthalmic problems, 56% having a convergent strabismus. Of patients reported in the literature, 10–15% have ophthalmic abnormalities including congenital blindness due to corneal opacity, congenital glaucoma, coloboma of the iris, choroid or optic nerve, convergent or divergent strabismus, nystagmus, cataracts and microphthalmia, ptosis, proptosis, medullated nerve fibres and retinal hamartomas.
Small keratin-filled cysts (milia) are found on the face in 30% of cases, most commonly in the infraorbital areas, but they can also occur on the forehead. Meibomian cysts on the corneal surface of the eyelids can cause great distress as they repeatedly discharge material. Skin tags are especially common around the neck; like the naevi, histology reveals the typical features of a BCC, but the skin tags do not generally change in size or shape.
Jaw Cysts
Some 13% of people with the syndrome develop a jaw cyst by the age of 10 years and 51% by the age of 20 years. The majority occur after the seventh year. The youngest affected person known to the author presented with jaw swelling at 5 years old. The peak incidence is in the third decade, about 10 years earlier than isolated odontogenic keratocysts. Around 26% of patients over the age of 40 years in the author’s series had not developed signs or symptoms of cysts; Gorlin gave a figure of 10% [8].
The mandible is involved far more frequently than the maxilla, with keratocysts occurring most usually at the angle of the mandible (Fig. 132.5).
Fig. 132.5 An orthopantogram of a 17-year-old male showing a large cyst in the angle and ramus of the left mandible.
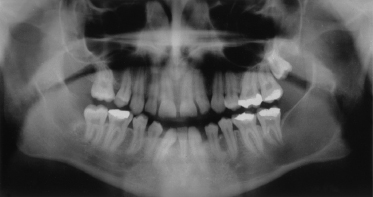
Patients can be remarkably free of symptoms until cysts reach a large size, especially when the ascending ramus is involved. Presentation can be with swelling and/or pain of the jaw, pus discharging into the oral cavity or displaced, impacted or loose teeth.
Mis-shapen teeth, missing teeth and a susceptibility to caries are more common in patients than in unaffected relatives.
Chest and Trunk
Epidermoid cysts occur on the limbs and trunk in over 50% of cases and are usually 1–2 cm in diameter. Rib anomalies may give an unusual shape to the chest, including a characteristic downward sloping of the shoulders (see Fig. 132.3). The rib anomalies together with kyphoscoliosis may cause pectus excavatum or carinatum in about 30–40% of patients. Sprengel deformity has been found in some surveys to be as common as 25%. Of males, 17% have inguinal herniae.
Hands and Feet
The distinctive pits found on the palms and soles appear to be pathognomonic [49]. They increase in number with age, are permanent and, when found in a child, are a strong diagnostic indicator. They may vary from only a few to over a hundred. BCCs have very rarely arisen in the base of the pits. In the author’s series, 65% had palmar pits by the age of 10 years, rising to 80% by the age of 15 years. They were present in 85% cases over the age of 20 years.
The pits are small (1–2 mm), often asymmetrical, shallow depressions, with the colour of the base being white, flesh-coloured or pale pink (Fig. 132.6). They are found more commonly on the palms (77%) than on the soles (50%). Pits can also appear independently on the sides of the fingers, when they are tiny, bright-red pinpricks.
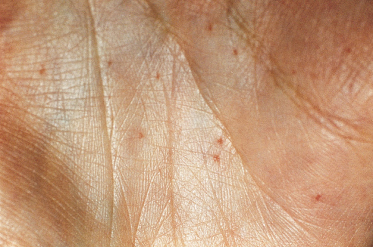
Although the pits will be easier to see in children who have been playing in a dirty environment, they should be differentiated from palmar lesions caused by excoriation of dirt under the skin. In most patients the pits can be better visualized if the hands are soaked in warm water for about 10 minutes.
Thumb anomalies (short terminal phalanges and/or small stiff thumbs) occur in about 10%. Pre- or postaxial polydactyly of hands or feet is found in 8%. The fourth metacarpal is short in 15–45% of patients, but is not a good diagnostic sign as it is found in about 10% of the normal population. Hallux valgus can be severe, requiring operation.
Naevi and Basal Cell Carcinomas
As ‘naevi’ and the BCCs found in the syndrome are histologically identical, they can both be classified as NBCCs. Clinically, however, the ‘naevi’ often develop first and behave differently from the BCCs, which can appear to arise from naevi. For the purposes of the clinical description that follows, it is helpful to treat ‘naevi’ and ‘NBCCs’ as though they are separate entities.
Naevi.
Naevi affect 53% of patients less than 20 years old, rising to 74% over the age of 20 years. Ordinary naevus cell naevi occur in about 4% of unaffected relatives but are present from birth, whereas affected family members report that the naevi tend to occur multiply in crops, their numbers increasing with time. Naevi also appear as individual lesions. A patient may develop no naevi, a few or many hundreds. The naevi are flesh-coloured, reddish-brown or pearly, the groups resembling moles, skin tags, ordinary naevus cell naevi or haemangiomas (Fig. 132.7). Some grow rapidly for a few days to a few weeks, but then most remain static.
Figs 132.7 Naevi. Note variation in size and appearance, some being reddish-brown, skin-coloured or translucent.
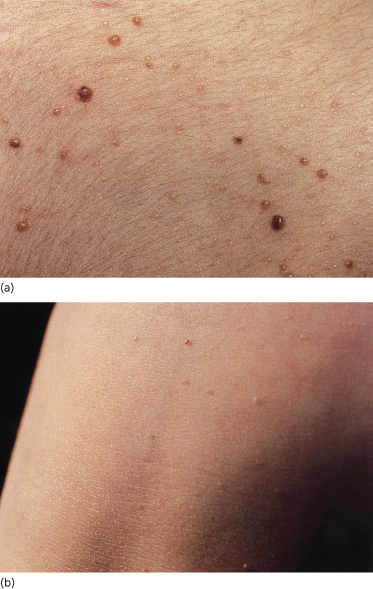
Although 53% of cases under the age of 20 years have naevi, only 14% present clinically in this age group with a rapidly growing BCC. It is unusual to develop aggressive BCCs before puberty.
Naevoid Basal Cell Carcinomas.
Basal cell carcinomas can arise in any area of the skin, affecting the face, neck and upper trunk in preference to the abdomen, lower trunk and extremities. The areas around the eyes, nose, malar regions and upper lip are the most frequently affected sites on the face. Usually only a few become aggressive, when they are locally invasive and behave like ordinary BCCs. Evidence of aggressive transformation of an individual lesion includes, as expected, an increase in size, ulceration, bleeding or crusting. Some patients can develop aggressive BCCs without first developing naevi. It is rare for metastasis to occur: only two cases have been reported [8].
In the author’s series, no children under the age of 10 years developed an NBCC requiring treatment. Of those aged 15 years, 20% had received treatment for one or more NBCC, 45% aged 20 years, 70% of those aged 25 years, 80% of those aged 40 years and 92% of those aged 45 years.
Skeletal Radiographs
Musculoskeletal features may be readily apparent on clinical examination. X-ray investigation may be helpful when the syndrome is suspected. Bifid, anteriorly splayed, fused, partially missing or hypoplastic ribs are found in 70% (Fig. 132.8
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


