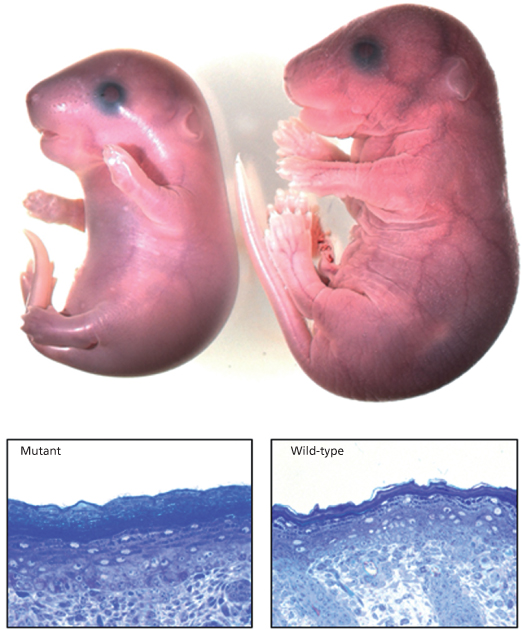
Fig. 13.2 (Upper panel) Mouse embryo with mutant ABCA12 is smaller than its wild-type littermate and has a shiny taut epidermis with limb contractures. (Lower panel) Mutant mouse epidermis displays hypergranulosis and marked hyperkeratosis compared with wild-type epidermis.
Courtesy of Ian Smyth, Monash University, Melbourne, Australia.
In vitro models of HI skin using human keratinocytes retrovirally transduced with shRNA targeting ABCA12 in a three-dimensional, organotypic co-culture system have also been developed by our group [24]. Depletion of ABCA12 expression had a dramatic effect, inducing premature keratinocyte differentiation and morphology comparable with that observed in HI skin, including a thicker epidermis and lipid dysregulation. Expression of the proteases kallikrein 5 and cathepsin D, associated with desquamation, was dramatically reduced in both HI epidermis and the organotypic co-culture (OTCC) model. These data suggest that ABCA12 is a key molecule in regulating keratinocyte differentiation and desquamation.
The majority of mutations causing HI are nonsense substitutions arising from an inserted premature termination codon, but deletions and insertions also may occur in the ABCA12 gene [25]. Founder mutations occur in different ethnic groups, e.g. 7322delC in the Pakistani population [26]. Compound heterozygotes may have a milder phenotype [27]. The majority of mutations can be identified using standard PCR and sequencing techniques but copy number molecular strategies including multiplex PCR or SNP array technology may be needed to identify complex deletions and insertions. To date, 95 of 100 HI cases tested in our laboratory have had mutations in ABCA12, suggesting that this is a homogeneous disorder genetically (Fig. 13.2). The five cases in which we have been unable to detect mutations may have a mutation within an intron, promoter or other regulatory component of the gene that has not been detected using conventional techniques. Uniparental disomy has also been reported [28].
Pathology.
Skin biopsy shows hypergranulosis, parakeratosis and massive, compact orthohyperkeratosis extending down into dilated hair follicles and pilosebaceous units. Usually, no other specific changes are noted on light microscopy and the basal and spinous layers appear relatively normal. In some cases, vacuoles are seen in the stratum corneum and the granular layer is reduced in thickness. Rarely, papillomatosis and a dermal inflammatory infiltrate are present. Pilosebaceous and sweat glands are preserved and non-keratinizing mucous membranes are normal.
Electron microscopy of 10 cases, including one fetal skin biopsy, revealed multiple vacuoles, lipid droplets and cellular remnants in corneocytes in all cases [11]. Lamellar bodies in spinous and granular cells were either absent or abnormal in all, and the intercellular lipid lamellae at the granular–corneal junction were not formed. Immunohistochemical staining of the cytoplasmic vesicles seen in granular layer cells suggested that they are abnormal lamellar granules [29].
In fetal skin biopsies, large abnormal mitochondria in keratinocytes have been noted at 16 weeks’ gestation, prior to keratinization [30]. Adnexal keratin plugging at 18–20 weeks preceded interfollicular changes, and a thickened cornified envelope was also evident.
Clinical Features.
The affected infant, usually premature, is encased in a grossly hyperkeratotic ‘coat of armour’ composed of large (2–5 cm), thick, yellow–brown, firmly adherent, dense plaques covering the whole body surface and severely restricting movement (Fig. 13.3). Soon after birth, the taut, inflexible cast splits, producing deep, red fissures extending into the dermis and a skin pattern resembling a harlequin’s costume. The facial features are distorted due to severe ectropion, conjunctival oedema (which obscures the eyes) and eclabium (open mouth and eversion of the lips). The scalp feels boggy and the nose and external ears are tethered and appear rudimentary with loss of the retroauricular fold. The oedematous hands and feet may either be encased in hard, mitten-like casts or covered with a mucoid membrane, but the digits are well formed underneath. Digital necrosis may occur, requiring surgical intervention. The skull may appear microcephalic. Additional congenital defects have been found in some cases.
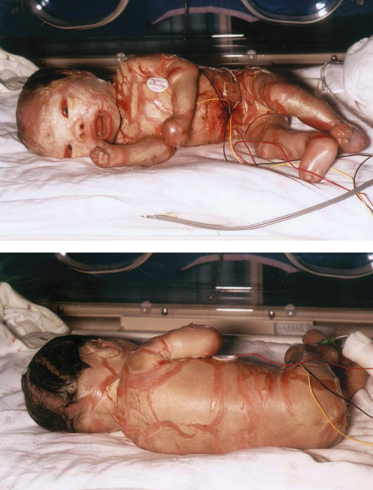
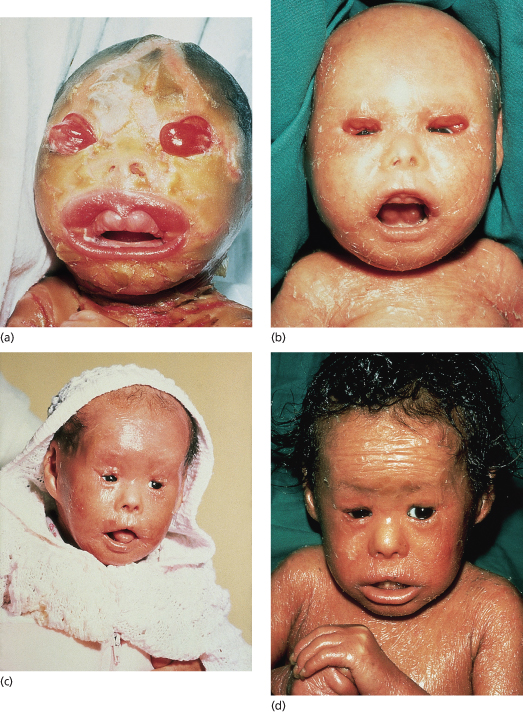
After shedding of the thick plates of skin, an erythrodermic ichthyosis resembling severe non-bullous ichthyosiform erythroderma is the eventual outcome. The patient reported by Lawlor [1,2] was the fourth of five affected babies, and the only survivor, in a kindred with nine other healthy children. She was commenced on etretinate in the first week of life and it was discontinued at 18 months (Fig. 13.4). At the age of 26 years she is growth retarded with severe ichthyosiform erythroderma and limb contractures but is well and attending mainstream college.
Fig. 13.4 Harlequin ichthyosis treated with etretinate at (a) birth, (b) 1 month, (c) 3 months and (d) 9 months.
Courtesy of Dr Sandra Charles-Holmes, Coventry.
Respiratory insufficiency resulting from restriction of chest movement may contribute to neonatal mortality. A defect in lung alveoli leading to alveolar collapse in the neonatal period is also plausible. In infants who survive the first days, the absence of effective sucking can cause feeding difficulties, leading to hypernatraemia, hypoglycaemia, dehydration and renal failure. Temperature instability and skin infection can complicate management. Intensive nursing and medical care, specialized incubators and ample fluid replacement from the outset, vigilance for infection, and early institution of oral retinoids are all factors which are resulting in increased HI survivors.
Prenatal Diagnosis.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


