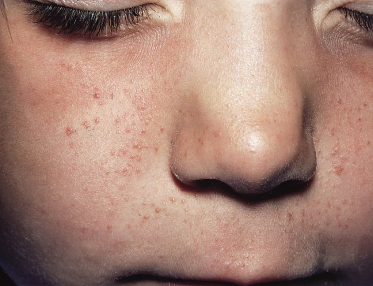
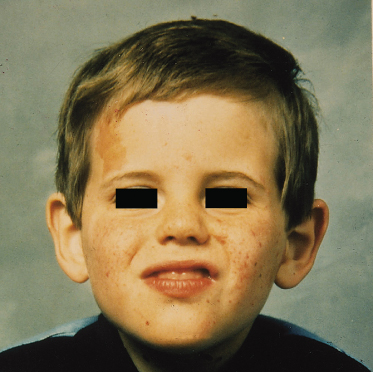
Fig. 129.3 Early facial erythema in a boy of 18 months who presented with infantile spasms in the first year of life. Facial erythema can precede the development of angiofibromatosis.
For a long time, the diagnosis was only considered in those with learning difficulties and the first attempts at an estimate of prevalence, in 1935, extrapolated the prevalence from the number of affected individuals in mental institutions [4] and suggested a figure of 1 in 30,000. A number of prevalence studies [5–8] have been undertaken since then, most of which suggest an overall prevalence of around 1 in 27,000, but with a much higher prevalence in children of nearer 1 in 10,000. This is supported by a long-term study in Minnesota, USA [9], suggesting a figure of 1 in 10,000 for a whole population. Because intellectually normal children with TSC may not come to medical attention, the birth incidence may be as high as 1 in 7000 [10]. Using capture–recapture techniques, a prevalence of 8.8 cases per 100,000 (1 in 11,363) was obtained in the UK but this has since been revised downwards to 5.1 per 100,000 (1 in 19,600) after a second ascertainment of the population.
Aetiology and Pathology.
Although TSC is an extremely variable condition, all variations of the disease would appear to occur within families, except when there is a deletion of the autosomal dominant polycystic kidney disease (ADPKD) gene in addition. Two genes seem to account for all cases of tuberous sclerosis. The first gene locus to be identified (TSC1) in 1987 showed linkage to the ABO blood group on chromosome 9q34 [11] and there is a second gene (TSC2) on chromosome 16 [12]. TSC2 was identified in 1993 [13,14], encodes for a protein named tuberin and is adjacent to the main gene for autosomal dominant polycystic kidney disease (ADPKD). The TSC1 gene was identified in 1997 and the protein product for TSC1 is called hamartin [15]. Proof that both the genes function as tumour suppressor genes came from the realization that hamartomas showed loss of heterozygosity for markers surrounding the TSC1 or TSC2 genes [16]. Effectively, both copies of the TSC1 or TSC2 gene in the hamartoma cell do not function; the first copy of the gene does not function due to an inherited mutation present in every cell, and the second copy is lost due to a spontaneous deletion of the second previously normal TSC allele. This spontaneous loss of the normal gene copy can be detected by looking for heterozygous markers in the normal chromosome within and around the TSC gene. When neither copy of the TSC gene functions in a cell, no TSC protein is produced and the tumour suppressor effect is lost. Additional evidence suggesting that tuberin is a tumour suppressor gene has been produced by using a tetracycline-responsive promoter to cause an overproduction of tuberin: these cells suffer from significantly inhibited cell growth.
The fact that the two TSC genes are tumour suppressor genes [17,18] has been shown to explain the clinical features of the disease. An affected individual starts life as an embryo with a single alteration in one copy of their TSC gene that was either an inherited or a spontaneous mutation. The latter may occur after the first mitotic division, leading to mosaicism. During normal growth and development of the fetus and later in life, in some cells in the body such as brain and cardiac muscle cells, a second random somatic mutation occurs, affecting the previously normal copy of the same TSC gene. These ‘second hits’ occur by chance during cell mitosis. When the affected cell has neither TSC gene functioning, it gives rise to the hamartoma that is the hallmark of the disease.
The Eker rat is an excellent example of a mendelian autosomal dominant predisposition to a specific cancer (renal carcinoma). A spontaneous germline insertion of repetitive DNA in the rat homologue of the human tuberous sclerosis (TSC2) gene causes to this abnormality in the Eker rat [18]. A tumour suppressor function of the TSC2 gene in the Eker rat has been demonstrated. Although the phenotype of TSC in humans differs from that in the Eker rat, renal carcinoma does occur infrequently in humans with TSC. Late in life, Eker rats may additionally develop haemangioscarcomas of the spleen, leiomyosarcomas of the uterus and adenomas of the pituitary. Neurological abnormalities have also now been shown. The rat TSC2 gene shares more than 90% of the amino acid pattern when compared with the human gene. It has also been shown that the introduction of the wild-type TSC2 gene can rescue the Eker rat from carcinogenesis.
It is now known that tuberin and hamartin combine together to function as a heterodimeric protein, explaining why loss of either gene causes the same basic phenotype. They are believed to function as a fine control mechanism within a cell growth pathway that is well known to cancer molecular geneticists. Tuberin and hamartin inhibit the mammalian target of rapamycin (mTor); rapamycin is a known immunosuppressive agent that has been used effectively to modulate immune function post renal transplant. mTor has effects on the S6K1 and P13K pathways and it has already been shown that rapamycin treatment of mice genetically engineered to have a TSC2 knockout can lead to resolution of the hamartomas. The mouse TSC2 knockout model may also show improvement in memory and learning deficits when treated with rapamycin.
Clinical Genetics.
Tuberous sclerosis complex is an autosomal dominant condition with a high spontaneous mutation rate of approximately 65%. The condition is fully penetrant, and non-penetrance is no longer thought to occur [6,8]. The diagnosis of tuberous sclerosis still remains a clinical one, and genetic testing in general is not necessary to make a diagnosis. However, genetic testing for mutations in either the TSC1 or TSC2 gene in the index case can be very helpful in managing the genetic implications of the condition for the wider family. Diagnostic testing for TSC1 and TSC2 mutations is now available, and a mutation in either gene can be identified in about 80% of either familial or sporadic cases. For the apparently healthy parents of a child with clinical TSC, if they have no cutaneous signs of TSC, it is unlikely that they are gene carriers and they can be offered confirmatory genetic testing if a TSC gene alteration is found in their child. If no gene alteration is identified in a child with clinical TSC, the parents may be offered detailed dermatological and ophthalmic clinical examination, followed by cranial imaging and a renal ultrasound scan. Cranial imaging for diagnosis used to be best carried out with a computed tomography (CT) scan to look for calcified subependymal nodules (Fig. 129.4) and this may still be sufficient in an adult. Cranial magnetic resonance imaging (MRI) is more likely to show tubers but the appearances are not always diagnostic and calcified subependymal nodules, which are diagnostic, can be missed on cranial MRI unless fluid-attenuated inversion recovery (FLAIR) sequences are performed (Figs 129.5 and 129.6).
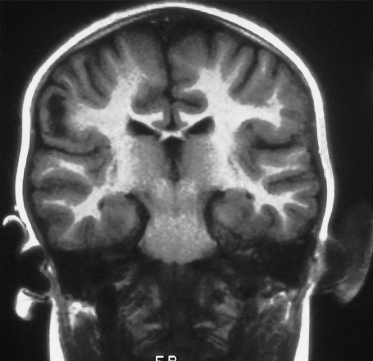
Fig. 129.4 This cranial CT scan shows multiple calcified subependymal nodules. In the left frontal region there is an area of reduced opacification, which represents a subcortical tuber; these are sometimes visible on CT scans.
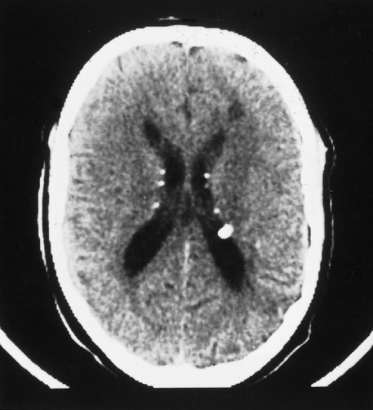
Fig. 129.5 This T2-weighted cranial MRI scan shows a large white area in the cortex of the left hemisphere, which is a cortical tuber.
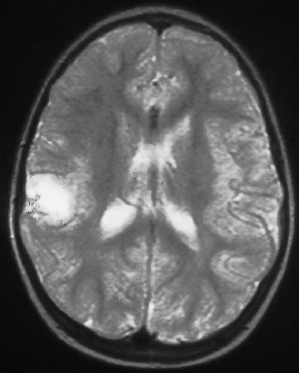
Fig. 129.6 This T1-weighted coronal–cranial MRI scan shows an ‘empty gyrus’ in the right parietal region, which is another imaging feature of a cortical tuber.
If all investigations are normal in a pair of parents, there still remains a risk of recurrence of approximately 2%, which is due to gonadal mosaicism. In gonadal mosaicism, a TSC gene alteration is present in either the ovaries or testes of mother or father, but there are no signs of TSC elsewhere, and no TSC mutation is found in their blood. Therefore, even if a child has a TSC gene mutation and neither of their parents is found to have that mutation in their blood, there still remains a 2% chance that those parents could have another child with TSC. For that reason, a number of parents choose to have genetic prenatal diagnosis for TSC in subsequent pregnancies, even when neither parent has their child’s TSC mutation.
Where a parent is affected by TSC, each child of an affected individual has a 50% risk of inheriting TSC, the severity of which is random (with the exception of polycystic renal disease) and cannot be predicted for an individual child. In general, disease caused by mutations in TSC1 is less severe than disease caused by mutations in TSC2. However, severe disease does occur due to TSC1 and mild disease also occurs in TSC2 [19].
If a TSC gene mutation is identified, siblings of an affected individual or children of an affected person should be offered a clinical genetics appointment to discuss predictive genetic testing, when they are old enough to understand the implications. Earlier investigation is only justified to exclude polycystic renal disease in the neonatal period when echocardiography may be contributory, when this evidence of TSC may subsequently disappear, or if symptoms require investigation. If no TSC mutation has been found in an index case, asymptomatic siblings should be offered a full screen (including cranial imaging) if their parents have not been examined, but a full dermatological and eye examination may be sufficient if the parents were found to be unaffected after full investigations.
Clinical Features
Neurological
Almost any organ in the body can be affected, with the possible exception of skeletal muscle and spinal cord, but it is the serious neurological sequelae that dominate the paediatric presentation. The most common scenario in approximately 50% of individuals is that of a young infant of between 3 and 7 months with infantile spasms. However, partial seizures, absence seizures (but not typical generalized absences) and tonic–clonic seizures are also found even in infancy. Seizures only occur in between two-thirds and three-quarters of patients. Of those with seizures, two-thirds will start their seizures in the first year but 10% will occur for the first time in school-age children and in 4% seizure onset is in adult life [8].
Learning disorder due to TSC is not thought to occur in the absence of seizures. The distribution of learning ability in individuals with TSC is bimodal: one-half will have an IQ in the normal range, whereas one-third will have profound disability, requiring supervision for daily living; often, these patients have little or no language but despite needing help with feeding, dressing and toileting, they are usually fully mobile. Little is known about the detailed neuropsychology of TSC. Clinically, there are sometimes areas of extraordinary ability in individuals with otherwise severe deficits and occasional severe localized deficits in individuals functioning normally. The possibility of a specific defect (using a test of visual discrimination and attentional set-shifting, the ID/ED shift) was recently reported in about one-half of individuals with TSC who have a normal IQ. The significance of this is not yet known. Autism, hyperactivity and sleep problems are the most frequent behavioural disorders but adults with TSC and IQ in the normal range have a high incidence of psychiatric disorders, including depression.
Up to 10% of infants presenting with infantile spasms will have TSC as the cause and all children with infantile spasms should have their skin examined, including with an ultraviolet light. This might also find other evidence of disease in this group of children, such as incontinentia pigmenti.
Focal neurological deficits also occur in TSC with a hemiparesis, usually mild, being most common. The cause of the hemiparesis can be an episode of status epilepticus or neurosurgery but in others is uncertain. Third nerve palsies have been seen. The head circumference is often slightly large and microcephaly should make one question the diagnosis, or consider two diagnoses.
Behaviour problems are one of the most distressing features of the disease. The combination of hyperactivity and autism is particularly difficult in children. Although these difficulties are more common in those with learning difficulties, they can occur in those with normal intellect who have had seizures. Improvement in behaviour is sometimes seen when better seizure control is achieved but it takes time to appear. In contrast with children with idiopathic autism, children with TSC and autism can still often learn from straightforward behavioural modification. Treatment of the hyperactivity is problematic, as until recently drug treatment was thought to be contraindicated by the presence of seizures. Some of the drugs used for the treatment of the seizures may themselves cause behavioural problems, usually aggression, especially clonazepam and clobazam; withdrawal of these drugs can produce dramatic improvements. Self-injury is often due to drugs or to frustration and poor communication, which can be relieved by attention to the detail of day-to-day life at home and at school. This can also result in some improvement of both behaviour and hyperactivity, occasionally dramatically so. Individuals in care homes need to be in a small home with only a few other occupants. They need to be kept occupied in a manner which they enjoy and behaviour problems then often reduce markedly.
Skin Manifestations
The earliest skin manifestations of TSC [20] include forehead plaques and shagreen patches as well as the more commonly recognized hypomelanic macules.
Forehead plaques may be present at birth (Fig. 129.7) or appear shortly afterwards and can initially look like a capillary haemangioma. They can occur anywhere over the scalp at the front of the face and neck. When fully developed they are a firm, fleshy, slightly raised lesion which is commonly red but can be yellow or brown.
Fig. 129.7 This large facial forehead plaque was noted when this boy developed infantile spasms: it was the earliest skin sign of TSC.
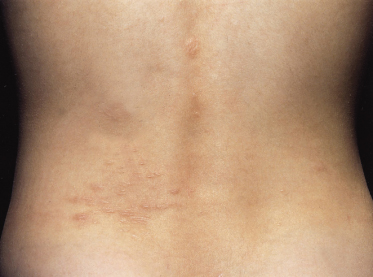
Shagreen patches when first developing may also look like a capillary haemangioma but there is usually some palpable thickening of the dermis. The typical site is posteriorly, in the lumbar region (Fig. 129.8), and occasionally at the top of the thigh. The shagreen patch is a roughened area of usually red but occasionally pale or slightly pigmented skin. Sometimes the appearance is similar to orange peel and the consistency is that of soft rubber. The patches may be only a few millimetres in size or extremely large, i.e. 10–15 cm in diameter. Large lesions sometimes have a crop of smaller lesions around them and occasionally only the smaller lesions are seen when they may be scattered over the back or localized in the more common area around the loin. Only a large classic shagreen patch should be considered a diagnostic feature. Histologically, again these are angiofibromas. They are rarely present in infancy but usually appear by adolescence. They are asymptomatic apart from their cosmetic appearance and are found in about 25% of individuals with TSC.
Fig. 129.8 This shagreen patch is at a typical site but is made up of multiple small areas of affected skin. The few small lesions distant from the main area are sometimes called satellite lesions.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


