NF1, neurofibromatosis type 1; NF2, neurofibromatosis type 2.
Neurofibromatosis Type 1
Epidemiology.
Neurofibromatosis type 1 is one of the most common autosomal dominant disorders, with an estimated prevalence of 1 in 3000 [1]. The birth incidence has been reported to vary between 1 in 2500 to 1 in 4000 [2]. NF1 affects all racial groups with no apparent predilection among any one particular group, although most epidemiological studies have involved mainly populations of European descent [3].
Diagnostic Criteria.
The National Institutes of Health (NIH) Consensus Development Conference on Neurofibromatosis published clinical diagnostic criteria for NF1 in 1988, which were reaffirmed unchanged in 1997 (Box 128.1) [4,5]. A diagnosis of NF1 can be confidently made after thorough clinical and ophthalmological examination in an individual when at least two of the features listed in Box 128.1 are present.
Box 128.1 NIH Diagnostic Criteria for Neurofibromatosis Type 1 (Two Are Required for Diagnosis)
1. Six or more café-au-lait macules (>5 mm pre-puberty, >15 mm post-puberty)
2. Two or more neurofibromas of any type, or one or more plexiform neurofibromas
3. Freckling in the axillae or groin
4. Optic glioma
5. Two or more Lisch nodules
6. Dysplasia of the sphenoid; dysplasia or thinning of long bone cortex
7. First-degree relative with NF1
These criteria have been shown to be both highly sensitive and specific for adults with NF1, but are less sensitive in children, particularly children under 8 years old, due to the fact that the number of clinical features increases with age. Forty six percent of sporadic NF1 cases fail to meet NIH diagnostic criteria by 1 year of age, but 95% of these children meet criteria by 8 years of age, and all do so by 20 years old [6]. The usual order of appearance of these features is multiple café-au-lait spots, axillary or inguinal freckling, Lisch nodules and neurofibromas. Characteristic osseous lesions are generally apparent within the first year and symptomatic optic gliomas are most often diagnosed by the age of four. Table 128.2 summarizes the characteristic age of onset of the diagnostic criteria.
Table 128.2 Percentage of patients with the cutaneous features of NF1 by age in a Welsh study [10]
Dermatological Features.
Three of the seven diagnostic criteria involve the skin; thus dermatologists play an important and often early role in the recognition and diagnosis of NF1. The major cutaneous features, café-au-lait spots, skinfold freckling and neurofibromas, will be reviewed in detail. Other cutaneous signs that have been reported to occur at an increased frequency in patients with NF1 will be briefly reviewed.
Café-Au-Lait Macules
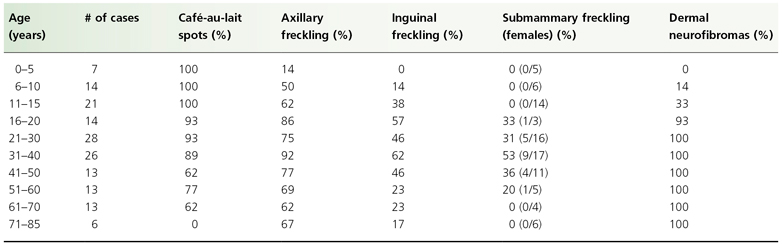
Café-au-lait macules (CALMs) are discrete, uniformly pigmented brown patches of varying sizes (Fig. 128.1). One to three café-au-lait macules are common in the population at large, occurring in up to 36% of children, but greater than three café-au-lait macules are significantly less common, occurring in only 0.3% of the general population and often indicate an underlying disorder, most commonly NF1 [7]. Crowe & Schull established six café-au-lait macules >1.5 cm as being useful in distinguishing those individuals with multiple CALMs and NF1 [8]. A prospective study evaluating the diagnostic outcome of children with multiple CALMs followed 41 children with at least six CALMs and 30 (73%) developed other signs of NF1 (24/41) or segmental NF1 (6/41) [9].
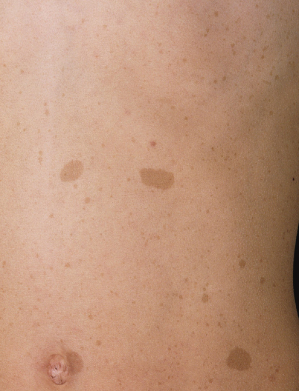
Café-au-lait macules are the earliest manifestation of NF1 and are present in essentially 100% of children with NF1. Conservatively, 80% of children with NF1 will manifest with multiple CALMs by the age of 1 [10]. If a child has not developed at least six CALMs by the age of 4, they are unlikely to do so [6]. CALMs continue to appear during childhood, but stop appearing or may disappear in adulthood. The spots vary in diameter from 0.5 to 50 cm or more, but are usually less than 10 cm. They increase proportionately in size with the growth of the child. The spots usually have smooth contours but some, particularly the larger ones, have irregular outlines. The colour intensity varies with background skin pigmentation, and in children with very pale complexions they are best seen under ultraviolet light.
Skinfold Freckling
Skinfold freckling (Crowe’s sign) is the most specific of the diagnostic criteria for NF1. It presents as multiple (>3) 1–3 mm tan macules in the axillae and inguinal folds (Fig. 128.2). Skinfold freckling is uncommon in infants and typically becomes evident around the age of 3, reaching a prevalence of 90% by the age of 7 [6]. In children with multiple CALMs and no family history of NF1, the appearance of skinfold freckling confirms the diagnosis of NF1. In addition to the axillae and inguinal folds, freckling can occur around the neck, in the submammary region, in other skinfolds and occasionally is a generalized finding in adult patients.
Neurofibromas
Neurofibromas are benign tumours of the nerve sheath, which can occur at any point along any peripheral nerve, from the spinal roots to the cutaneous free nerve endings. Neurofibromas can be broadly classified as focal (localized to a single site on a nerve) or ‘plexiform’. Dermatologists will most frequently encounter focal cutaneous and subcutaneous neurofibromas and superficial plexiform neurofibromas.
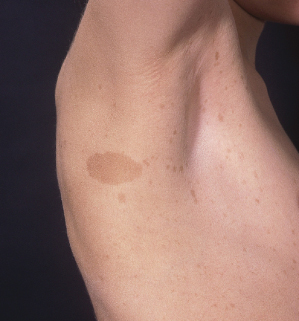
Cutaneous neurofibromas are generally first noticed in adolescence and continue to increase in size and number throughout adulthood. Younger children may have neurofibromas that appear as subtle subcutaneous swellings that are best seen with side lighting. Almost all adults with NF1 will develop cutaneous neurofibromas [10]. The number an individual will ultimately develop is unpredictable, even among family members, and can range from a few to thousands of lesions. Women with NF1 often report an increase in size and number of neurofibromas during pregnancy. They appear as soft, dome-shaped, flesh-coloured to slightly hyperpigmented papules or nodules or as more subtle bluish lesions that barely project above the skin surface (Fig. 128.3). When pressed, the tumours tend to invaginate into the subcutaneous tissue, a sign called buttonholing [8]. Cutaneous neurofibromas do not have a risk of malignant degeneration, but have a negative impact on quality of life due to disfigurement from the visibility and sheer number of lesions [11]. Cutaneous neurofibromas can be pruritic and occasionally, especially if large and pedunculated, infarct, which can present with pain and swelling.
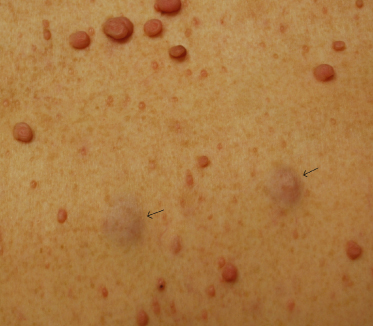
Subcutaneous neurofibromas are less prevalent than cutaneous neurofibromas and arise along peripheral nerves under the skin. These lesions present as well-defined, ovoid, firm subcutaneous nodules, best appreciated with palpation. Subcutaneous neurofibromas are often painful and can cause neurological symptoms, such as paraesthesia or weakness. While the risk of malignancy in these tumours is low, it is important to recognize individuals with multiple subcutaneous tumours because they are more likely to have internal spinal or plexiform neurofibromas, which are at a higher risk for malignant change [12]. The presence of subcutaneous neurofibromas has also been reported to be associated with a higher mortality rate [13].
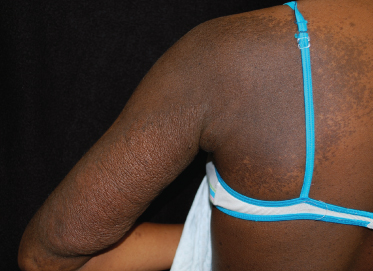
Plexiform neurofibromas (PNFs) are neurofibromas that develop along the length of the nerve and involve multiple nerve fascicles and even multiple branches of nerves. The term ‘plexiform’ arises from the histopathological appearance and implies a network-like growth of neurofibroma involving multiple nerve fascicles. Plexiform neurofibromas may be superficial and visible on examination, or deep and apparent only with imaging. Superficial plexiform neurofibromas are usually noticed early in life. A population study in Wales found that 27% of individuals with NF1 had a plexiform neurofibroma apparent on examination and in another study up to 44% had a plexiform neurofibroma apparent with imaging [10,14]. When they are superficial, there is often associated overlying hyperpigmentation, hypertrichosis and/or increased vascular markings (similar in appearance to a capillary malformation) (Fig. 128.4). The skin appears thickened and there are palpable cord-like masses, often likened to a ‘bag of worms’. Overlying irregular hyperpigmentation is sometimes the earliest clue to an underlying plexiform neurofibroma and can appear before the skin thickens. Plexiform neurofibromas can be associated with soft tissue overgrowth, leading to massive hypertrophy and disfigurement. When plexiform neurofibromas involve the face they can cause facial asymmetry and there is often associated dysplasia of the greater wing of the sphenoid [15]. Unlike cutaneous neurofibromas, there is a risk of plexiform neurofibromas transforming into malignant peripheral nerve sheath tumours (MPNST). The estimated lifetime risk of MPNST in individuals with NF1 is 8–13% [16]. Unrelenting pain, sudden growth or increased firmness in a previously stable PNF are all signs suggestive of malignant transformation and should be evaluated. Positron emission tomography (PET) and PET computed tomography have been shown to be useful for evaluating malignant transformation in symptomatic plexiform neurofibromas [17].
Other Dermatological Associations
Juvenile xanthogranulomas (JXGs) occur at an increased frequency in the NF1 population and the triple association of JXGs, NF1 and the development of juvenile myelomonocytic leukaemia has been reported [18–21]. The exact frequency of the triple association is unknown and a source of debate [22,23]. JXGs have a similar appearance and natural history in patients with and without NF1. Routine haematological screening in patients with NF1 and JXGs is currently not recommended, but clinicians should be aware of the possibility of juvenile myelomonocytic leukaemia and be vigilant for its presenting features (hepatosplenomegaly, lymphadenopathy, pallor, petechiae).
Generalized hyperpigmentation has been noted in individuals with NF1 compared to their unaffected siblings or parents [24]. Interestingly, the involved body regions of patients with segmental NF1 often have a background of hyperpigmentation. Although the cause of the generalized hyperpigmentation has not been studied, it is interesting to theorize that it is caused by a single mutation of the NF1 gene, whereas CALMS are caused by loss of both alleles.
There are several case reports of multiple subungual glomus tumours occurring with NF1 [25–27]. Patients presented with severe pain localized to one or more fingers and cold intolerance. It is important to recognize the increased risk of glomus tumours in NF1 patients because surgical removal of the tumour eliminates the pain.
Non-Dermatological Features.
Neurofibromatosis type 1 can involve any organ system. Reported non-cutaneous manifestations are summarized in Table 128.3. The non-cutaneous features that comprise the diagnostic criteria, those that may be recognized during physical examination, and learning disabilities are reviewed.
Table 128.3 Non-cutaneous manifestations and typical period of presentation
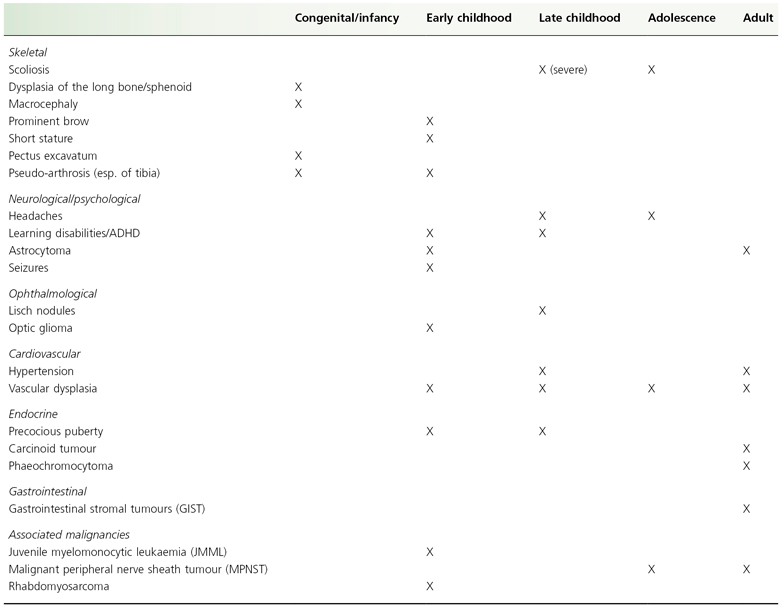
ADHD, attention deficit hyperactivity disorder.
Orthopaedic
Orthopaedic manifestations are frequent and include mildly short stature, skeletal dysplasia, scoliosis and more recently recognized osteopenia/osteoporosis. Sphenoid wing or long bone dysplasias are the most distinctive bony lesions in NF1. Approximately 14% of patients with NF1 fulfil this diagnostic criterion, which is usually evident by the age of 1 [6]. Long bone dysplasia should be suspected clinically when an infant or child presents with anterolateral bowing of the lower leg or, less commonly, the distal arm. These children are at an increased risk for fractures, which often fail to heal normally and can lead to pseudo-arthrosis. Contrary to the wording of the NIH criteria, thinning of the long bone cortex is uncommon, and the characteristic radiographic finding is cortical thickening with medullary canal narrowing [28]. Sphenoid wing dysplasia presents as facial asymmetry with either enophthalmos or proptosis, depending on whether there is associated orbital plexiform neurofibroma. If physical examination does not suggest a bony abnormality, routine radiographs are not recommended. Scoliosis is also common, with an incidence of 10–25% [29]. Although the dermatologist’s role is limited, periodic screening of children and adolescents is simple and if evidence of scoliosis is found, referral to orthopaedics is warranted.
Ophthalmological
Lisch nodules (iris hamartomas) are 1–2 mm smooth dome-shaped lesions on the iris [30]. They can occasionally be detected on general examination as yellow-brown lesions, but are best viewed and distinguished from iris naevi with slit-lamp examination. Lisch nodules are innocuous, but are useful for diagnostic confirmation of NF1. They generally appear after the CALMs and skinfold freckling and are found in approximately 40% of children under 6, 85% of children under 18 and 93% of adults [31,32].
Optic pathway gliomas are classified as low-grade pilocytic astrocytomas of the optic nerve, optic chiasm, hypothalamus or optic tracts. Optic pathway gliomas are estimated to occur in 15% of children [33]; one-third to one-half of these cause clinical symptoms, such as decreased visual acuity, diminished visual fields, proptosis or precocious puberty [34]. Optic pathway gliomas are slow to progress and many never require intervention. Symptomatic optic pathway gliomas usually manifest by the age of 6, but there are an increasing number of reports of late-onset or late-progressive optic pathway gliomas in NF1 [35]. The 1997 OPG Task Force determined that there was not enough evidence to recommend routine neuroimaging in asymptomatic children. Instead, children with known or suspected NF1 should have yearly ophthalmological assessments until at least the age of 7 [36].
Learning Disabilities
Learning disabilities are the most common complication in children with NF1. The reported frequency of learning disability ranges between 30% and 69% [37]. Several studies have shown a consistent shift to the left of IQ but it is within 1 SD of the population mean; mental retardation (IQ <70) is uncommon [38–41]. Learning problems in children with NF1 can be global learning deficits (with associated lowered IQ) or specific learning deficits, with a normal IQ but lower than expected academic achievement. Children with NF1 have particular difficulties with visuospatial orientation, receptive and expressive language skills, and gross and fine motor co-ordination [42] The learning problems are compounded by an increased prevalence of attention deficit hyperactivity disorder (ADHD); 46 of 93 patients in one series satisfied the criteria for ADHD [43].
Pathology.
Only the pathology of café-au-lait macules, neurofibromas and plexiform neurofibromas will be reviewed. For other tumours that occur, the underlying pathology is usually the same as when they occur in isolation [44].
Café-Au-Lait Spots
Microscopically, café-au-lait spots present as focal hyperpigmentation in the basal layer of an otherwise normal epidermis; axillary freckles show a similar appearance. There is an increase in dopa-positive melanocytes, which show an increased number of melanin macroglobules [45,46]. Melanocytes with melanin macroglobules are also found, though in much lower numbers, throughout the skin of NF1 patients. Macroglobules are not specific to NF1 and are an occasional finding in normal individuals and other disorders including McCune–Albright syndrome, LEOPARD syndrome and xeroderma pigmentosum [47].
Neurofibromas
Histologically, neurofibromas in patients with NF1 are indistinguishable from those occurring as isolated lesions in the general population. Cutaneous neurofibromas are well circumscribed but not encapsulated, and although they are thought to arise from the terminal branches of cutaneous nerves, the nerve of origin is not usually obvious. Conversely, subcutaneous neurofibromas develop on major peripheral nerves, are more sharply demarcated and have an apparent capsule of perineural cells. The fibres of the nerve of origin pass through subcutaneous neurofibromas and are not stretched over their surface, as is seen in schwannomas [48].
Microscopically, both dermal and subcutaneous neurofibromas are spindle cell tumours originating from peripheral nerve sheaths [48,49]. They are composed of five cell types including Schwann cells, neurones, fibroblasts, perineurial and mast cells. Common features include the formation of collagen fibres and myxoid degeneration. Mitotic activity is low. The tumour is not locally invasive. Cutaneous neurofibromas are not thought to have the potential for sarcomatous degeneration.
Plexiform Neurofibromas
Plexiform neurofibromas are classically thought to always occur in association with NF1 but they can occasionally be seen as isolated lesions in otherwise healthy individuals and presumably they then represent somatic mosaicism. Plexiform neurofibromas can be discrete nodular tumours developing on a nerve root or diffuse tumours associated with hypertrophy of surrounding connective tissue and other structures [49]. The tumour cells are embedded in an abundant extracellular myxoid matrix, which often contains mast cells. Plexiform neurofibromas are locally invasive, growing within and along the nerve, enlarging nerve fascicles and elongating each fascicle. In the early stages of the lesion, hypercellular fascicles are found. As the lesion develops, there is an increase in the number of Schwann cells and/or perineurial cells. A few residual axons that have not been destroyed by the tumour can be found. As the lesion grows, the fascicle can become either hypocellular and myxomatous or even more cellular; the two pathologies can be found side by side in a single lesion. Plexiform neurofibromas have the potential for malignant degeneration into sarcomas [48,49].
Pathogenesis
Genetics
Neurofibromatosis type 1 is inherited as an autosomal dominant trait. Approximately one-half of affected individuals have a de novo mutation and therefore have no family history of NF1. Penetrance approaches 100% by the age of 20, but expression is extremely variable, even within families [50]. This point is important for genetic counselling, because an individual with mild clinical findings can have a child with a more severe phenotype, or vice versa.
NF1 Gene and Neurofibromin.
The NF1 gene was mapped to chromosome 17 by linkage analysis in 1987 [50,51] and cloned in 1990 [52–55]. The gene is large, spanning 350 kb of genomic DNA, encodes an RNA of 11–13 kb and consists of 60 exons [56]. The NF1 gene encodes a protein called neurofibromin, which contains 2818 amino acids and has an estimated molecular mass of 220 kDa [57]. Neurofibromin contains a guanosine triphosphatase (GTPase)-activating protein (GAP) domain. This domain has been demonstrated to be active and is the only part of the protein whose function is known [58]. GAP proteins convert Ras from its active GTP-bound form to its inactive GDP-bound form, thus suppressing cell proliferation. The presence of this domain supports a tumour suppressor function for the NF1 gene.
The cutaneous manifestations of NF1 have been shown to result from mutational events in accordance with Knudson’s two-hit hypothesis of tumour formation, further evidence to support the tumour suppressor function of neurofibromin. Several studies have shown that cutaneous neurofibromas develop only after both copies of the NF1 gene are lost [59–61]. Notably, it is only a subpopulation of Schwann cells that harbour the somatic (second-hit) mutation and not the other tumour cells (e.g. fibroblasts, mast cells), suggesting that the Schwann cell is the tumour cell of the neurofibroma [62]. It is hypothesized that the subpopulations of Schwann cells with two mutated NF1 alleles secrete growth factors with subsequent recruitment and proliferation of other cells [63]. The somatic mutations in neurofibromas are distinct from the germline (first-hit) mutation and different for each neurofibroma [62]. Café-au-lait macules have also recently been shown to result from loss of both NF1 alleles. DeSchepper and colleagues found second-hit NF1 mutations in melanocytes cultured from café-au-lait macules; second-hit mutations were not present in fibroblasts, keratinocytes or melanocytes from uninvolved skin [64].
The large size of the gene and the lack of mutational hotspots have made mutation analysis challenging. Messiaen et al. developed a comprehensive multistep detection protocol that is able to identify mutations in greater than 95% of patients (both familial and sporadic) who fulfil NIH criteria [65]. Pathogenic mutations have been found in most of the 60 exons and comprise a wide diversity of mutation types, including complete gene deletions, chromosome rearrangements, smaller deletions or insertions, stop mutations, amino acid substitutions and splicing mutations. Indeed, most mutations result in absent or non-functional protein [66].
Genotype-Phenotype Correlations.
Genotype-phenotype correlations have been difficult to establish because of the complexity of both the phenotype and the gene, but there are two of interest to dermatologists. Individuals with total loss of the NF1
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


