
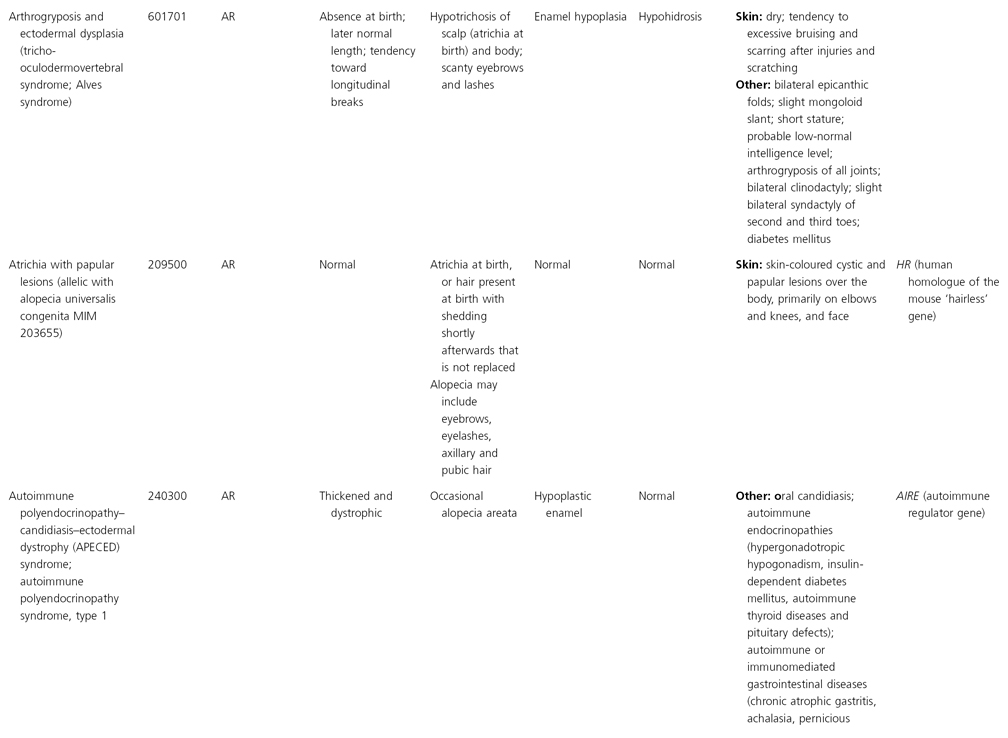
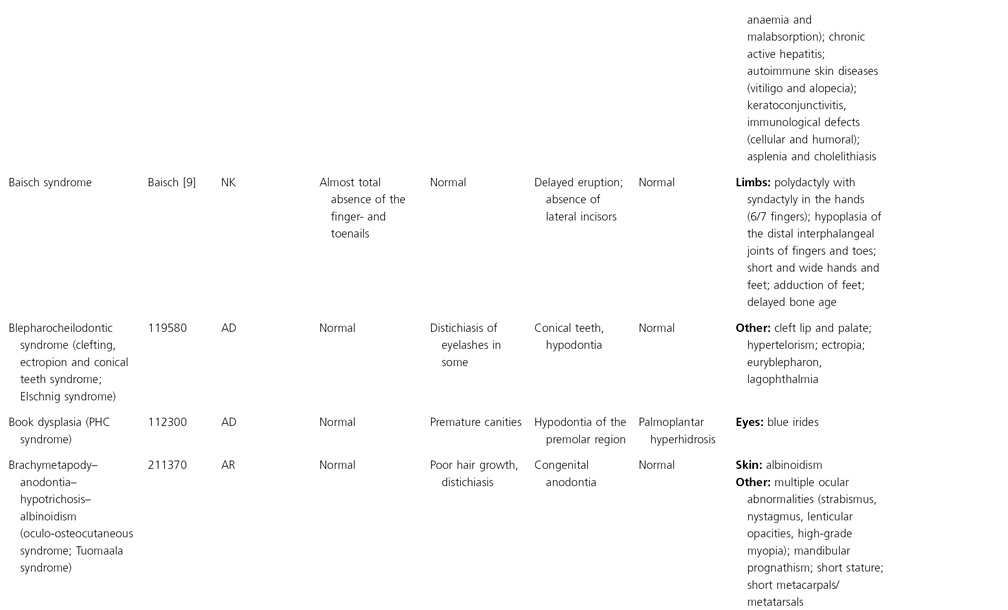
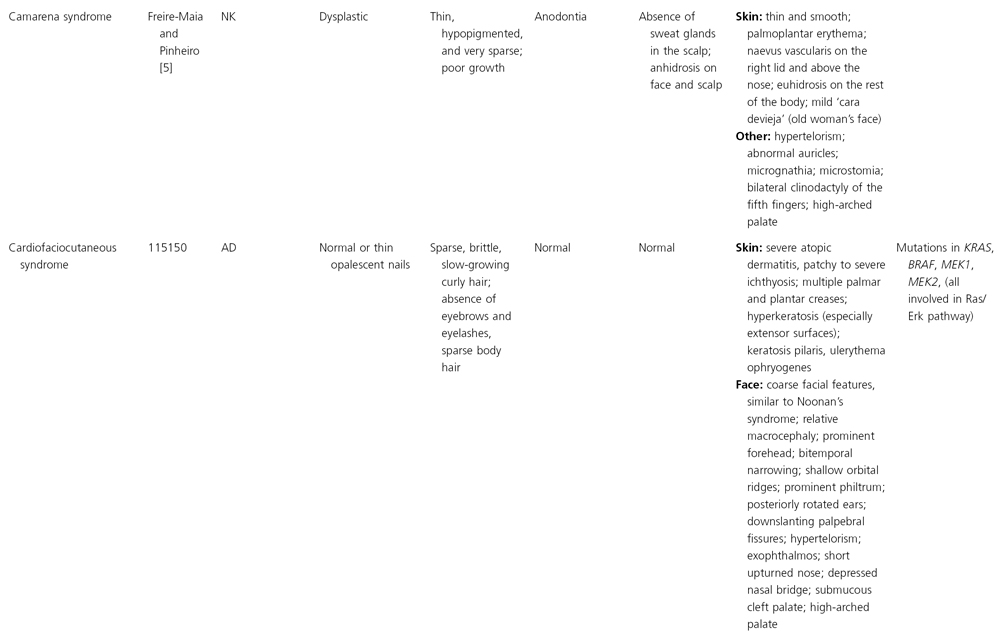

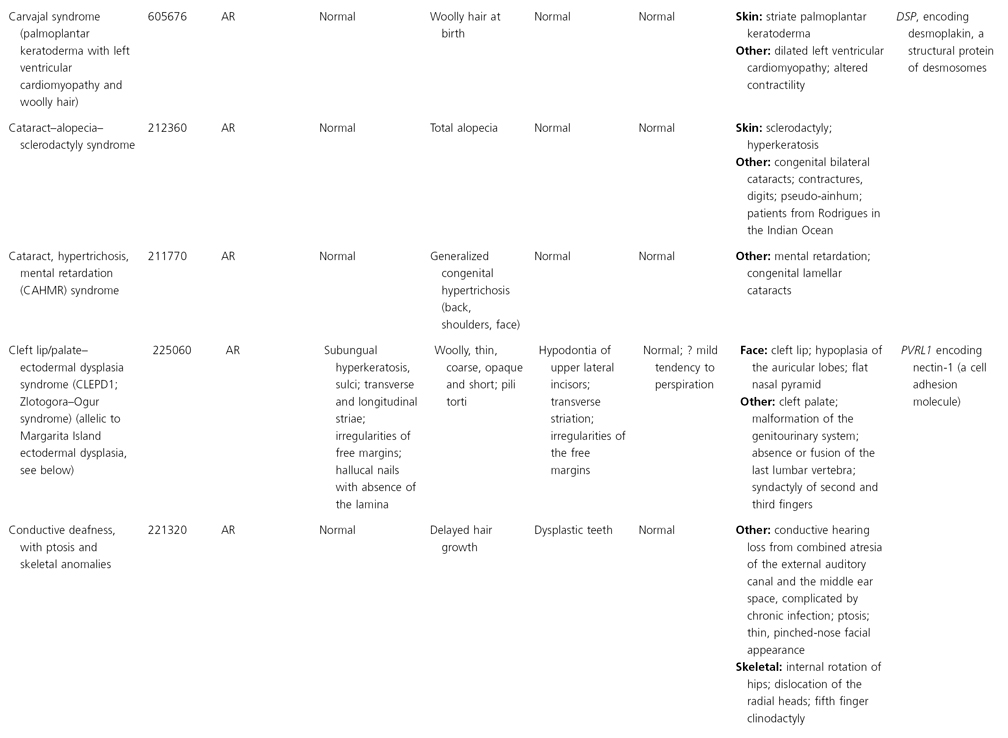
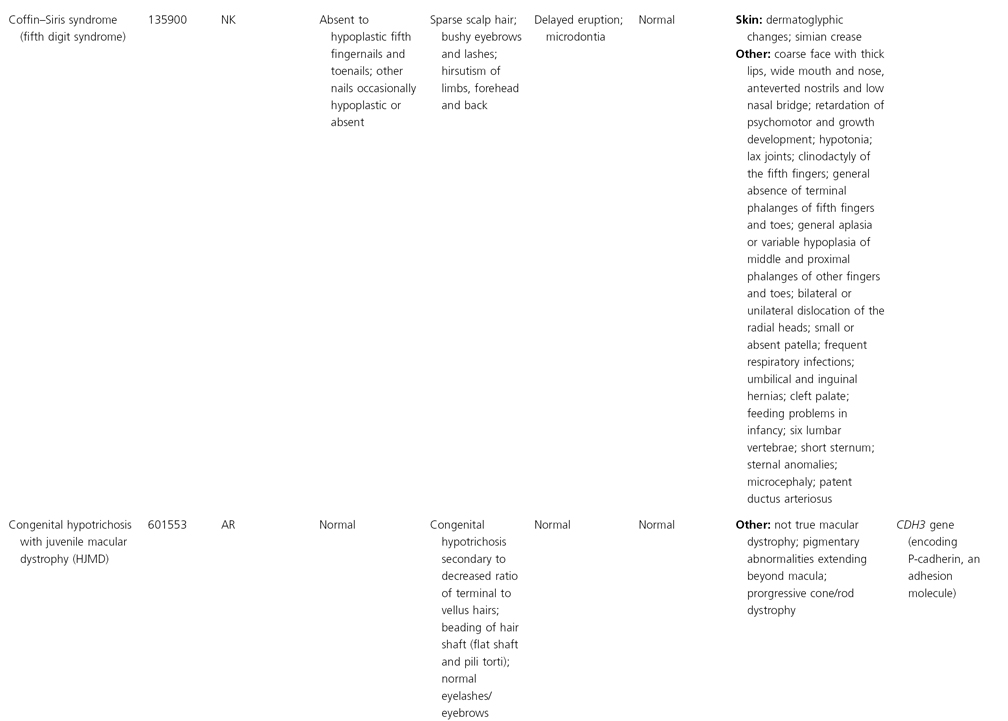
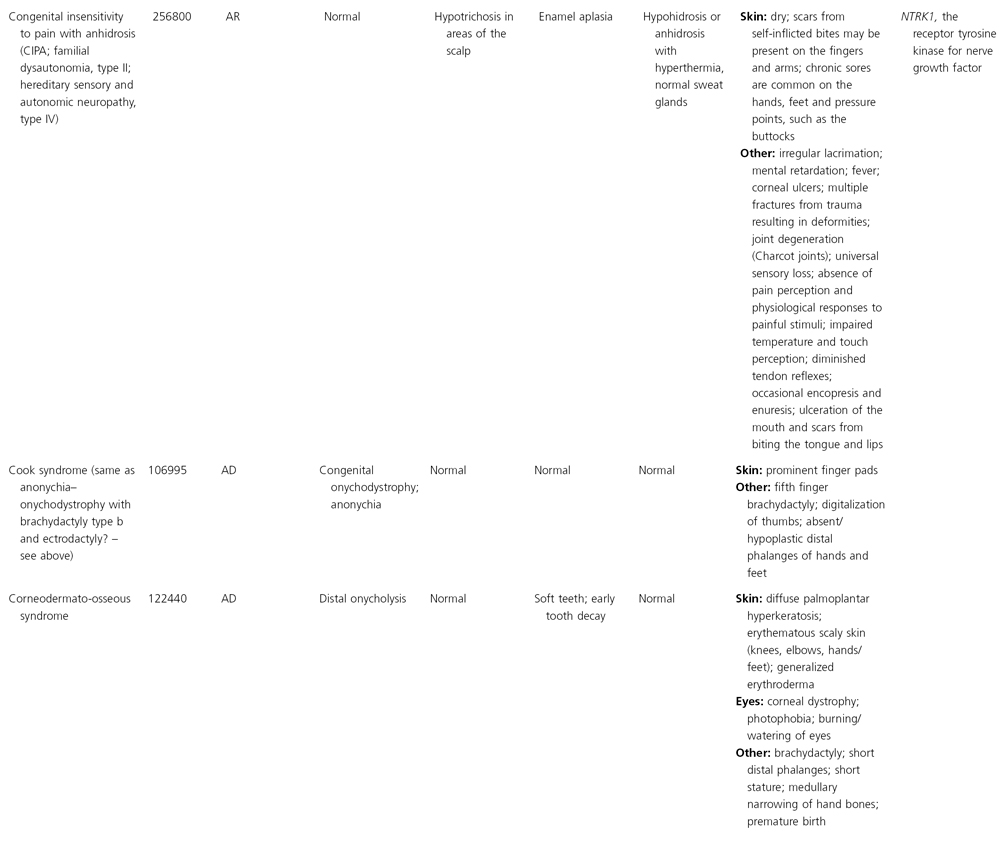
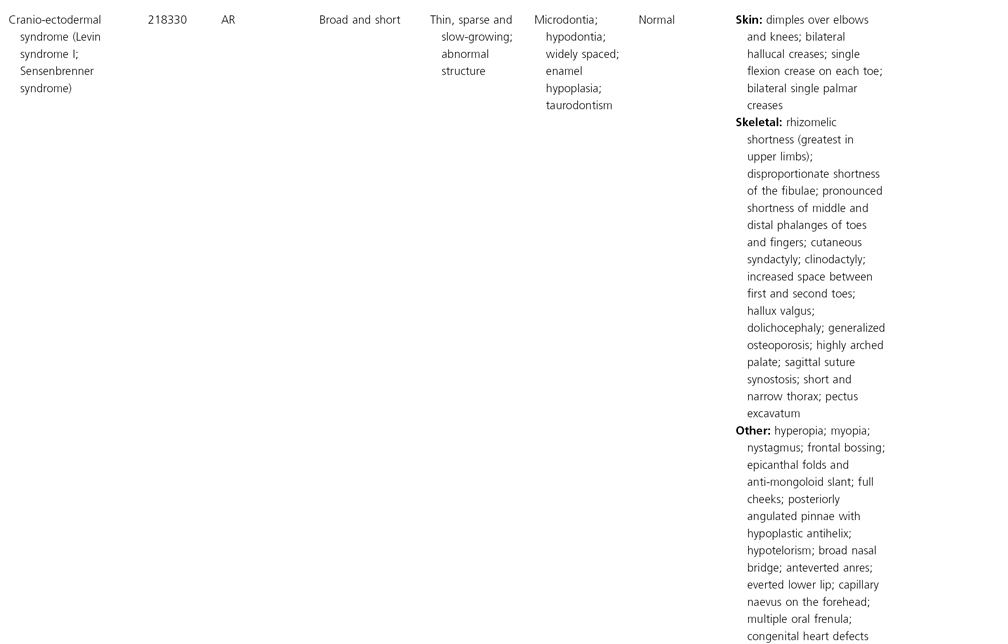
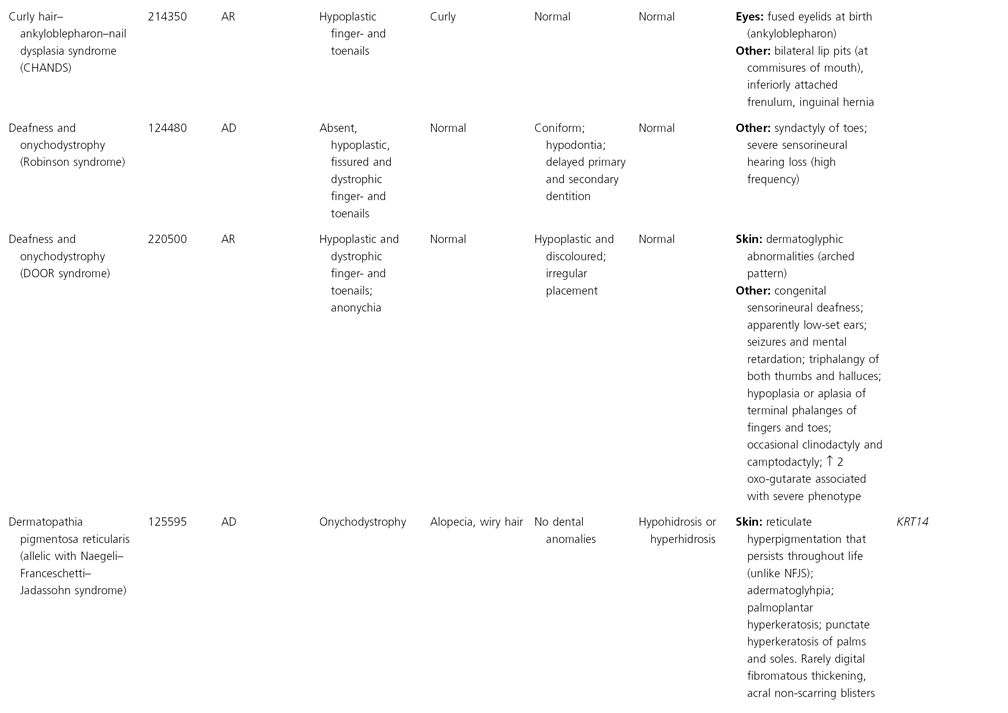

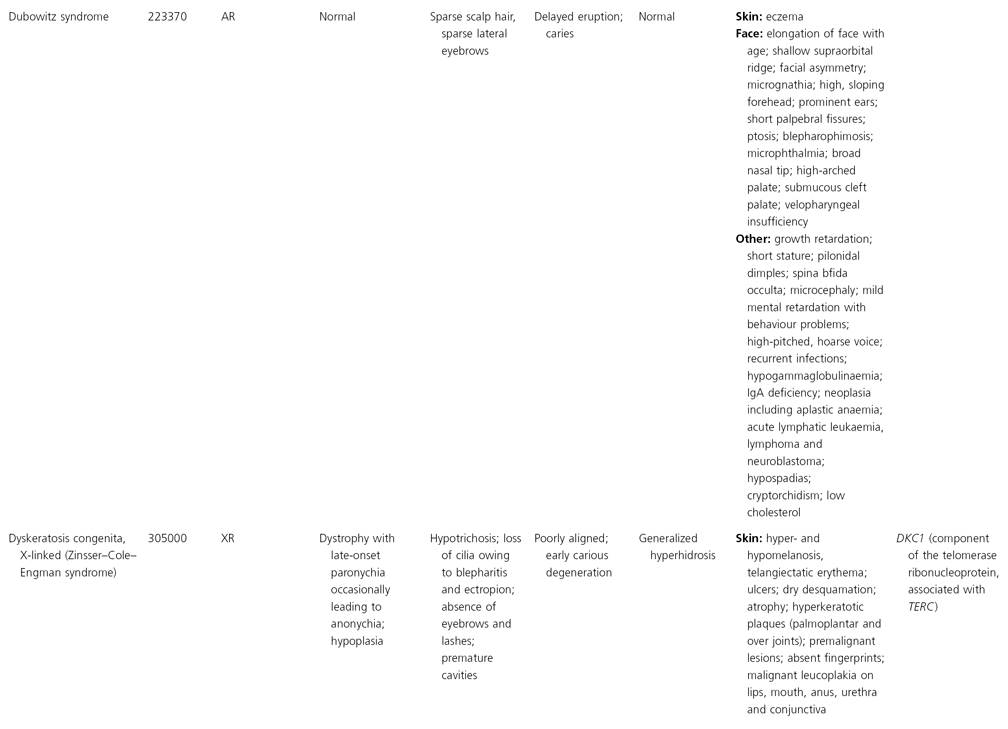

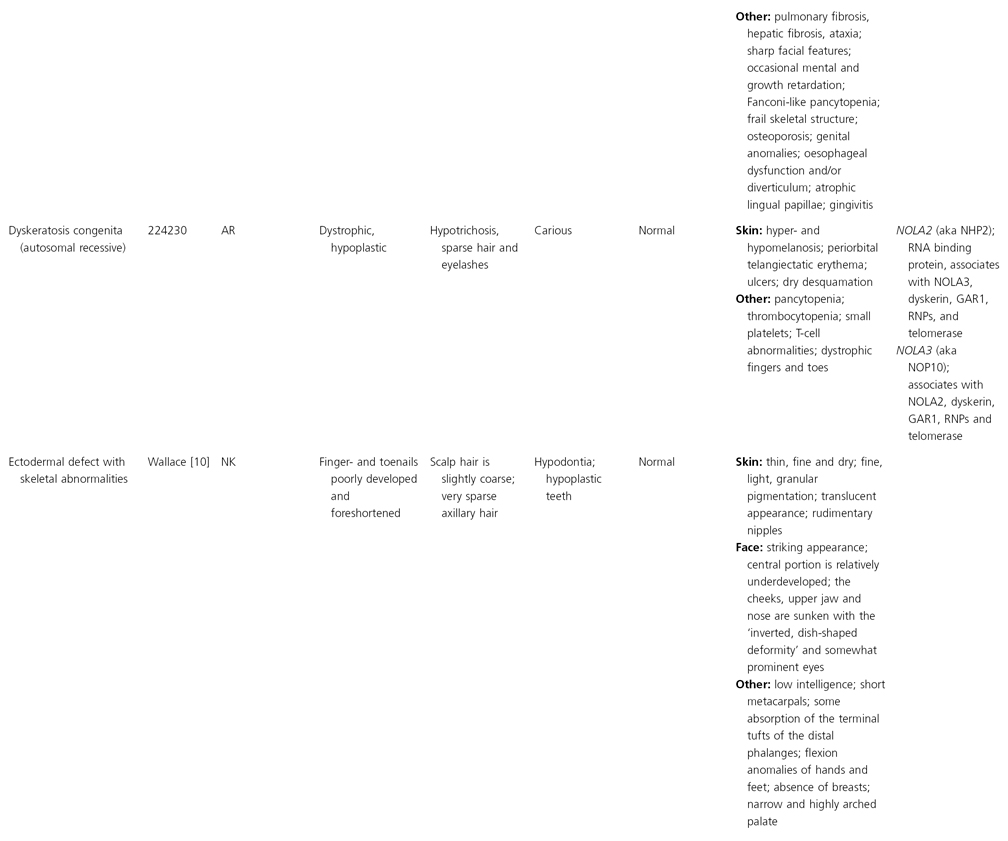
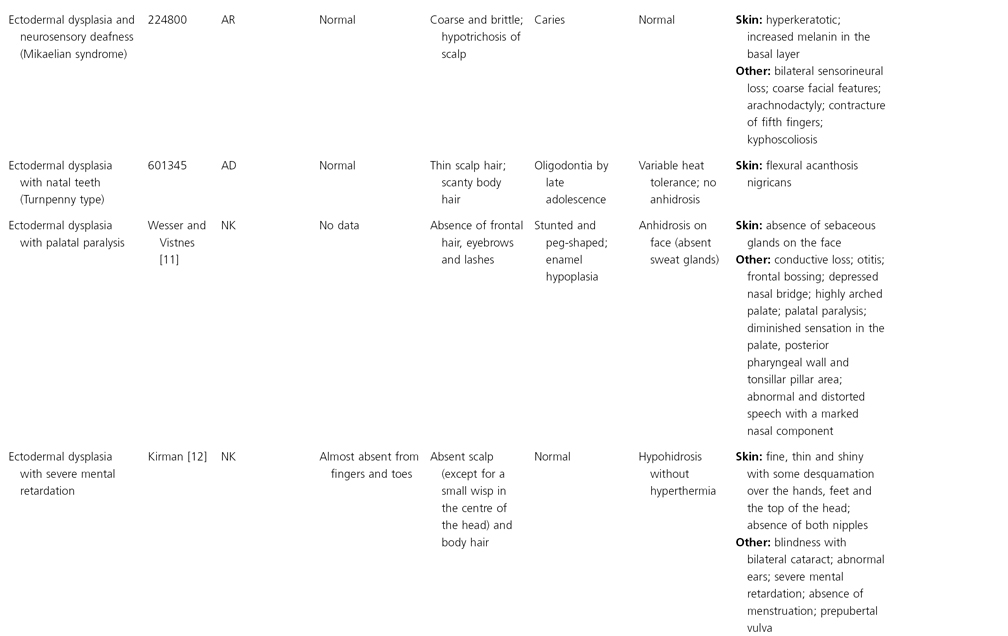
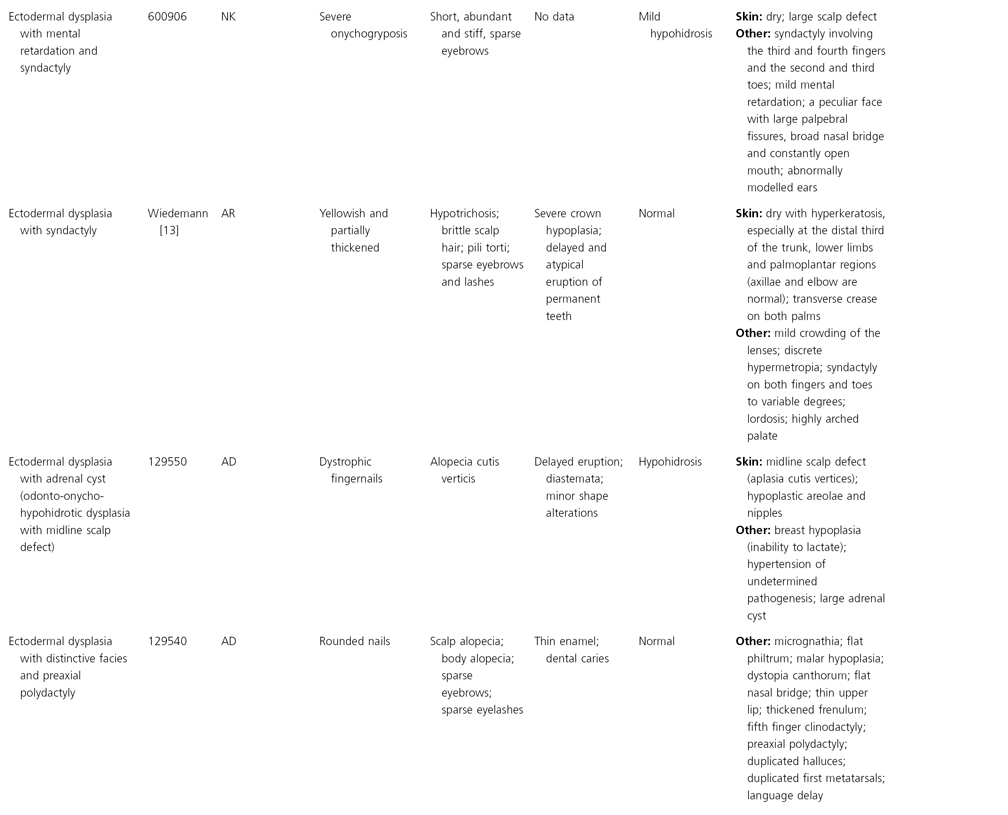
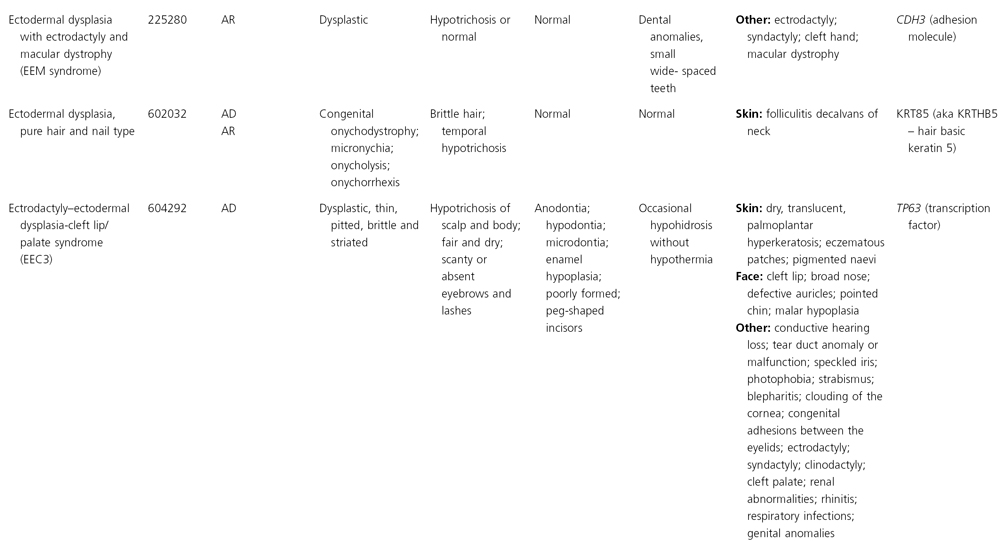
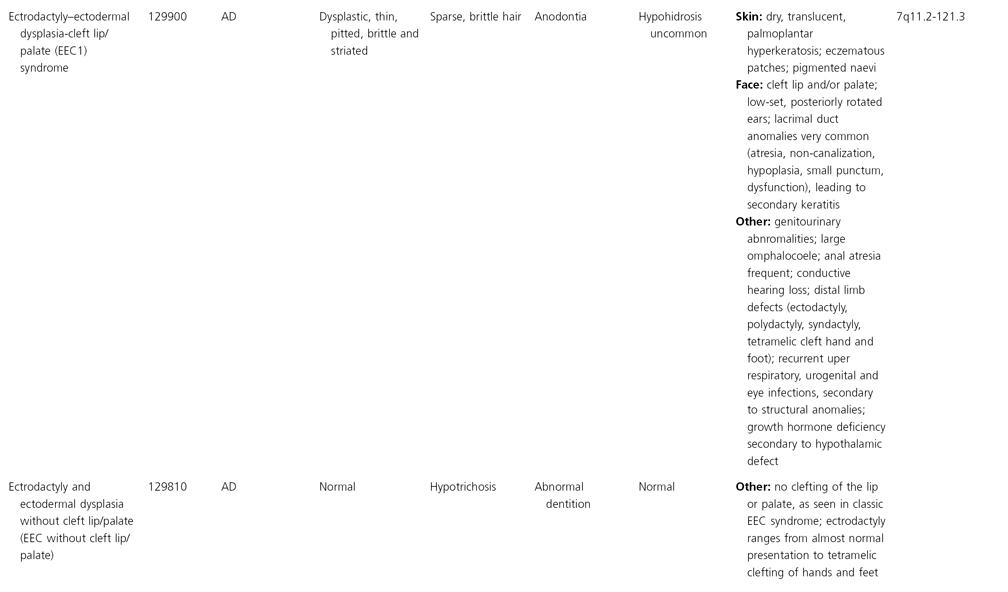
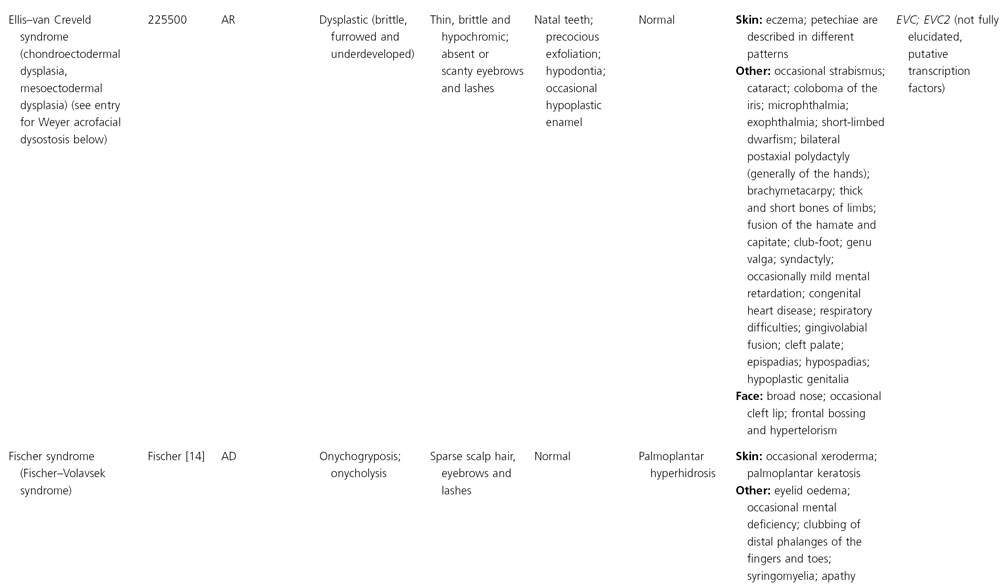
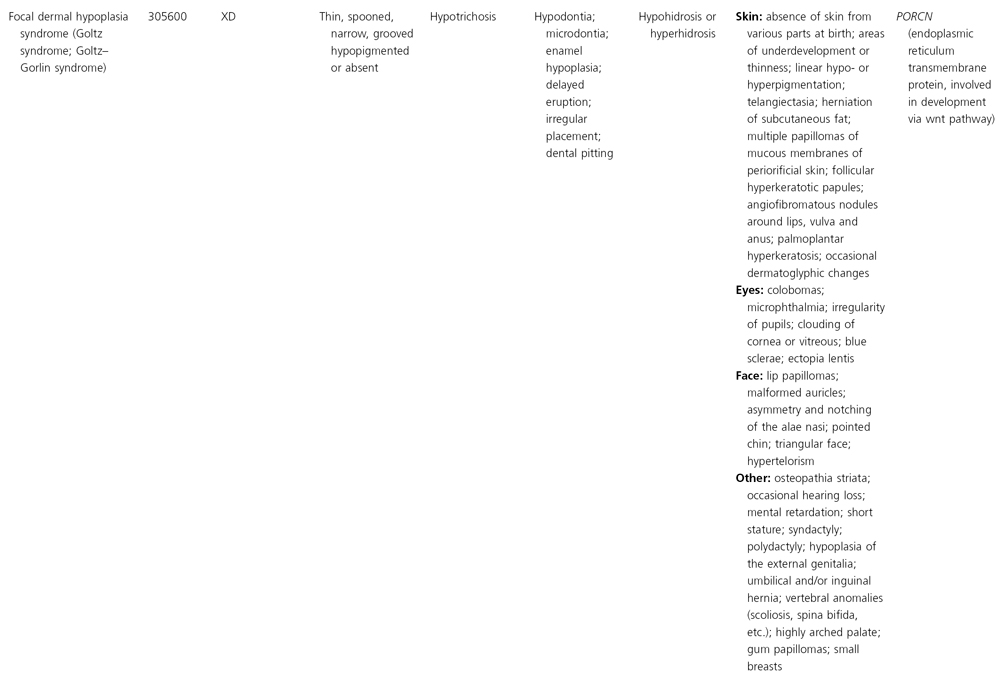
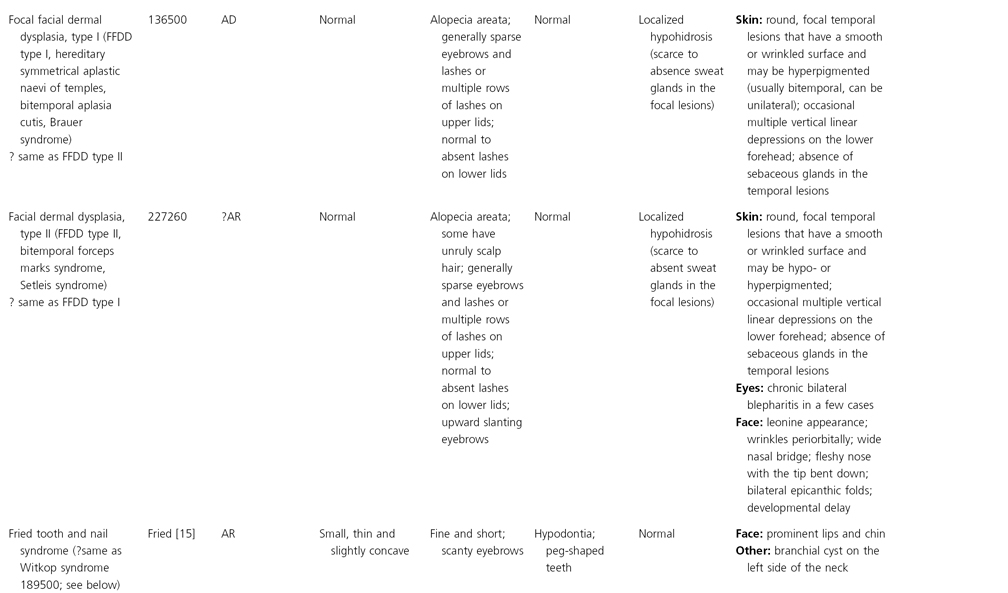
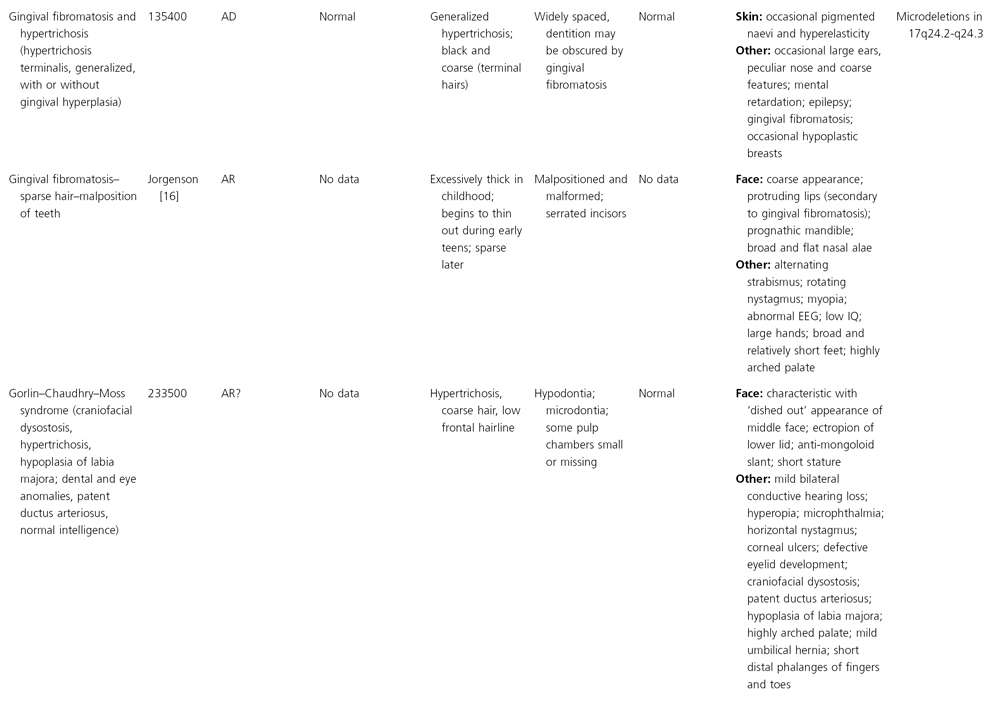
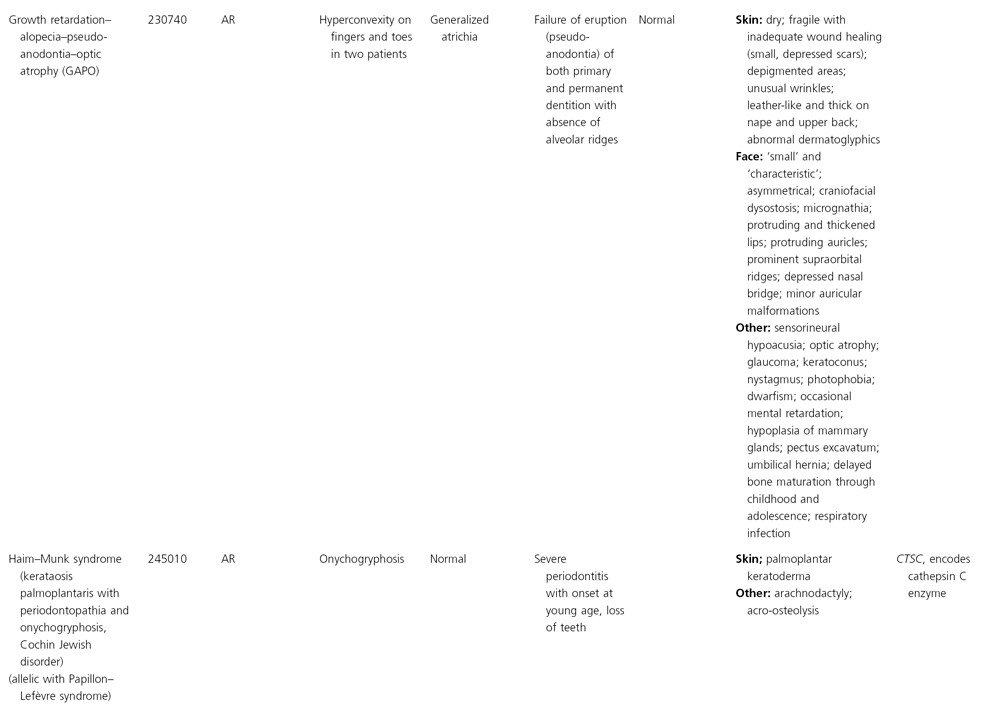
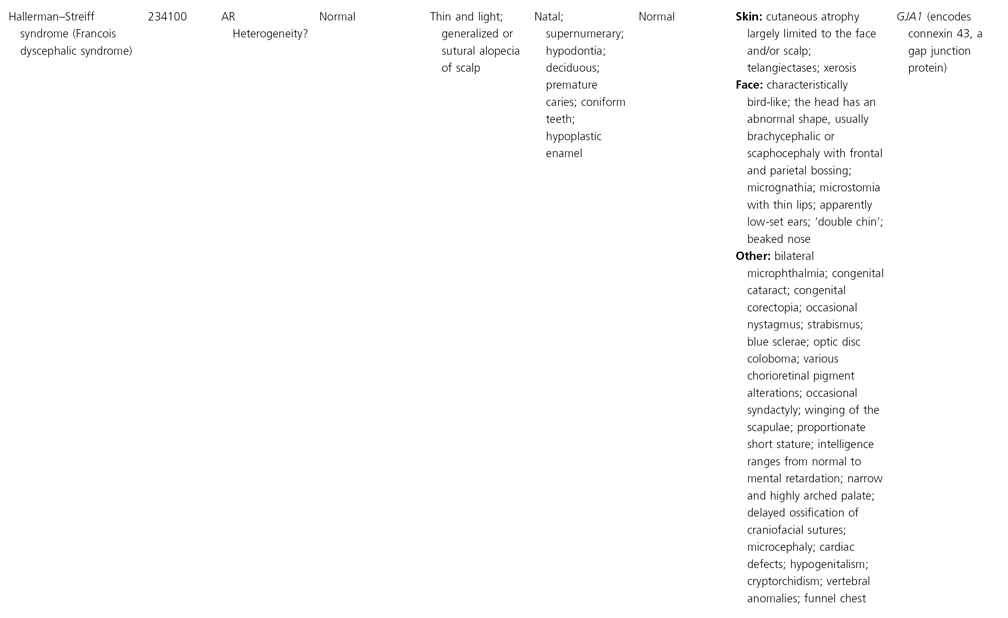
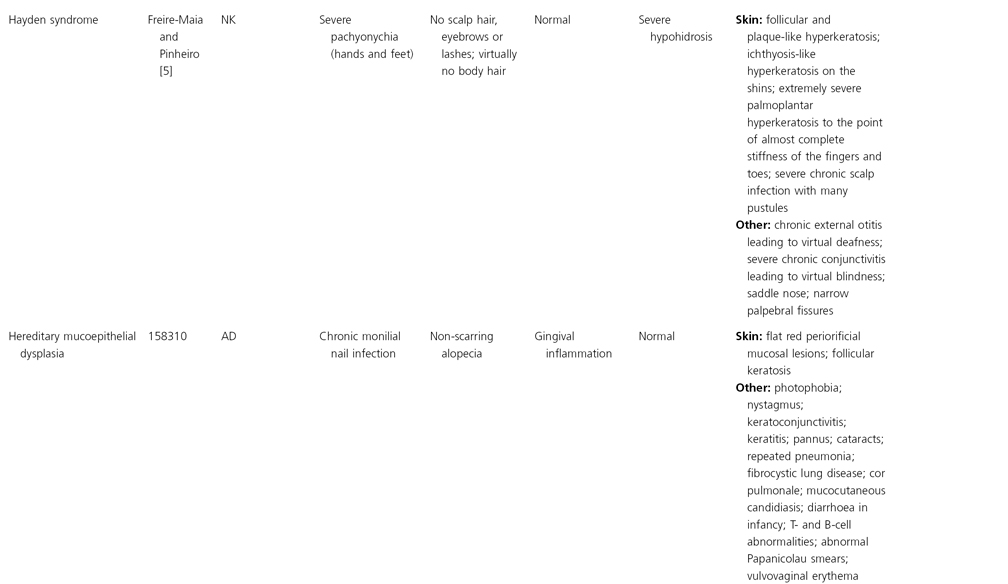
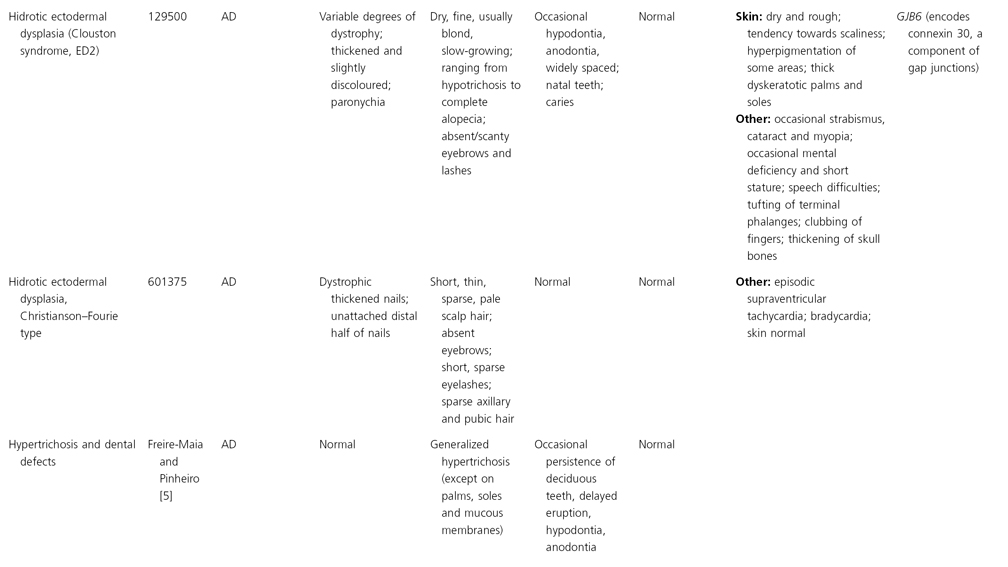
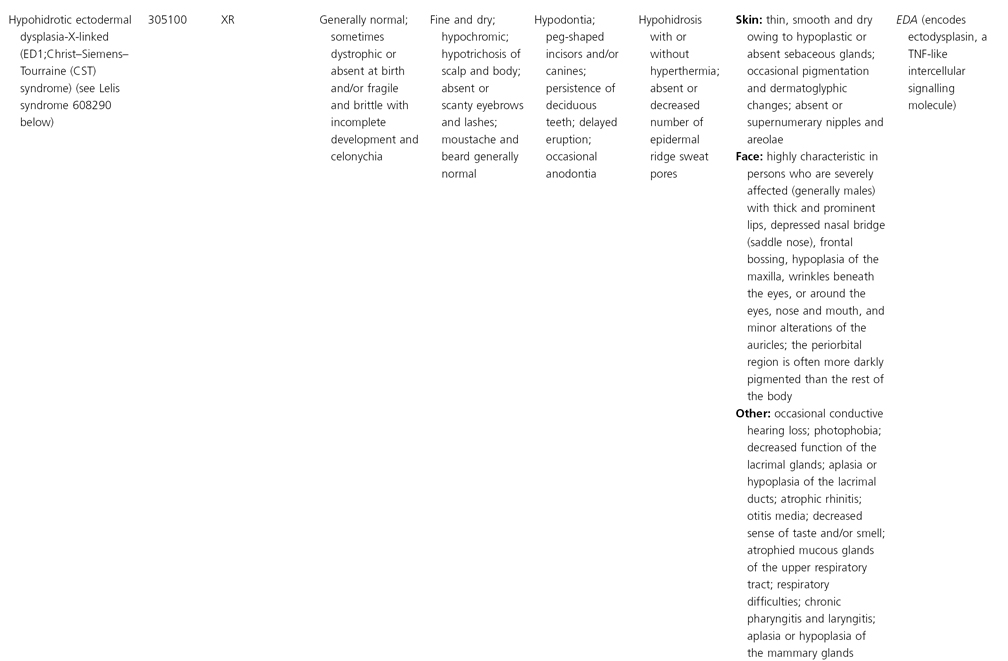
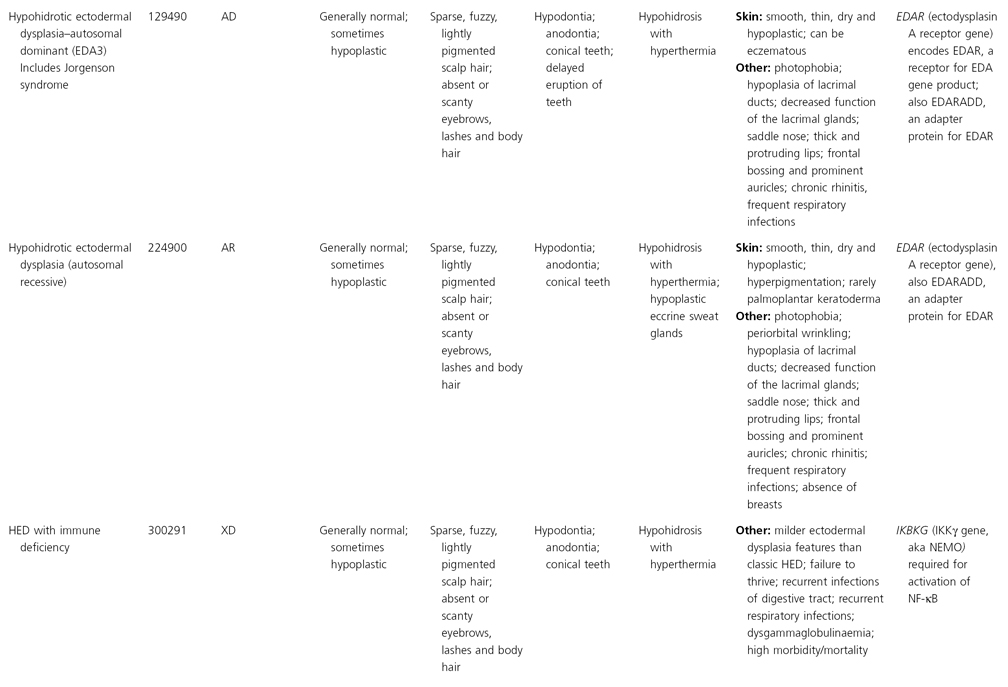
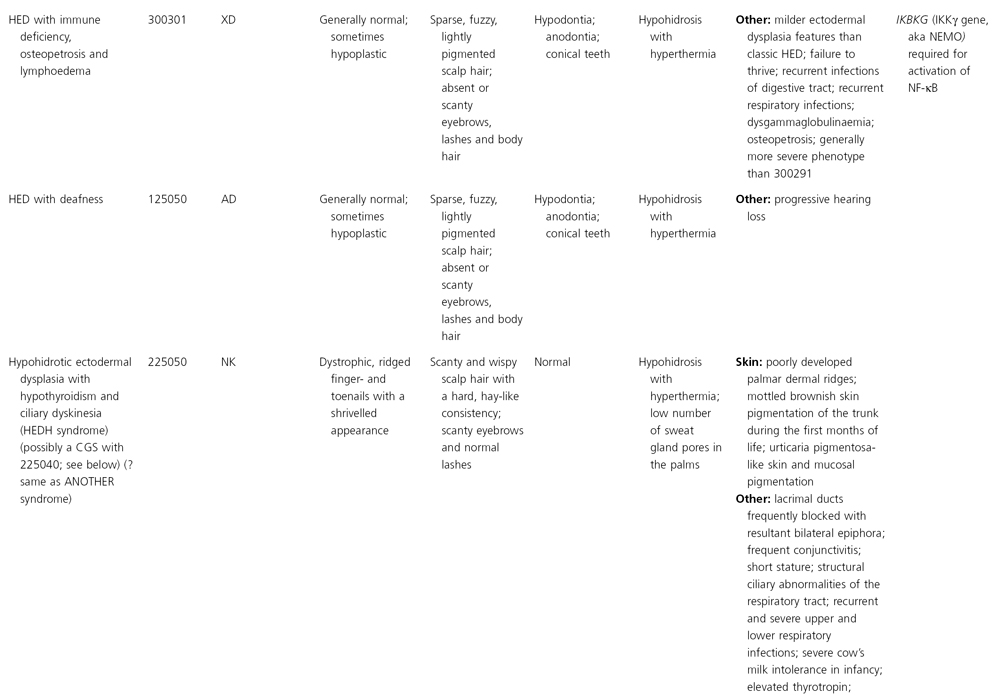
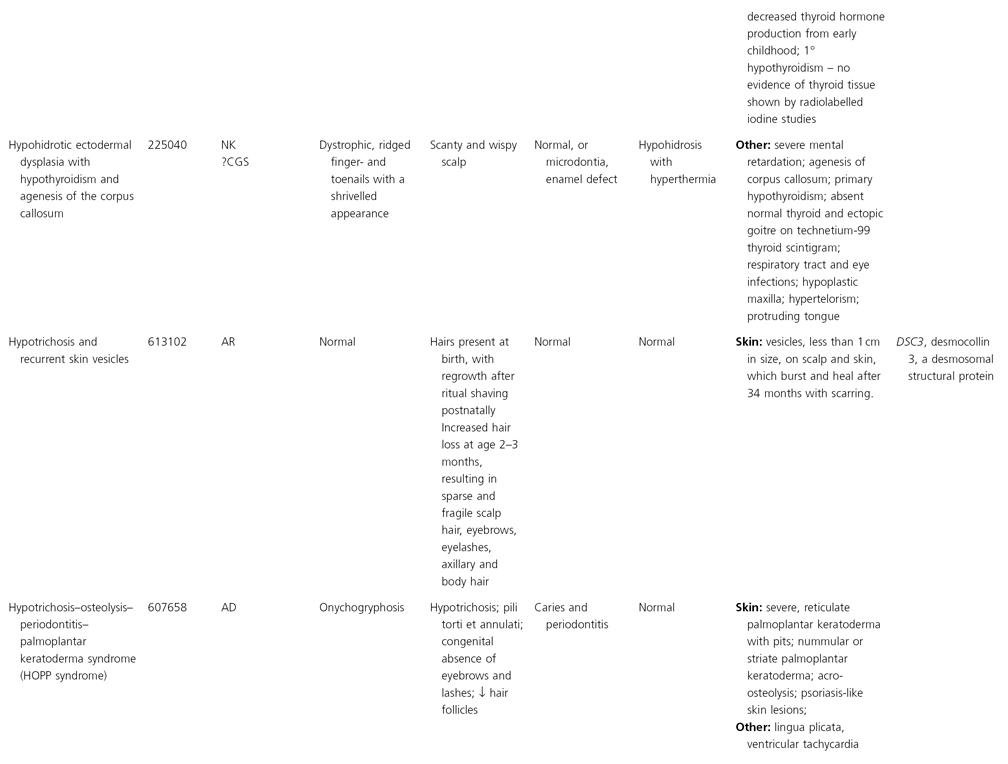
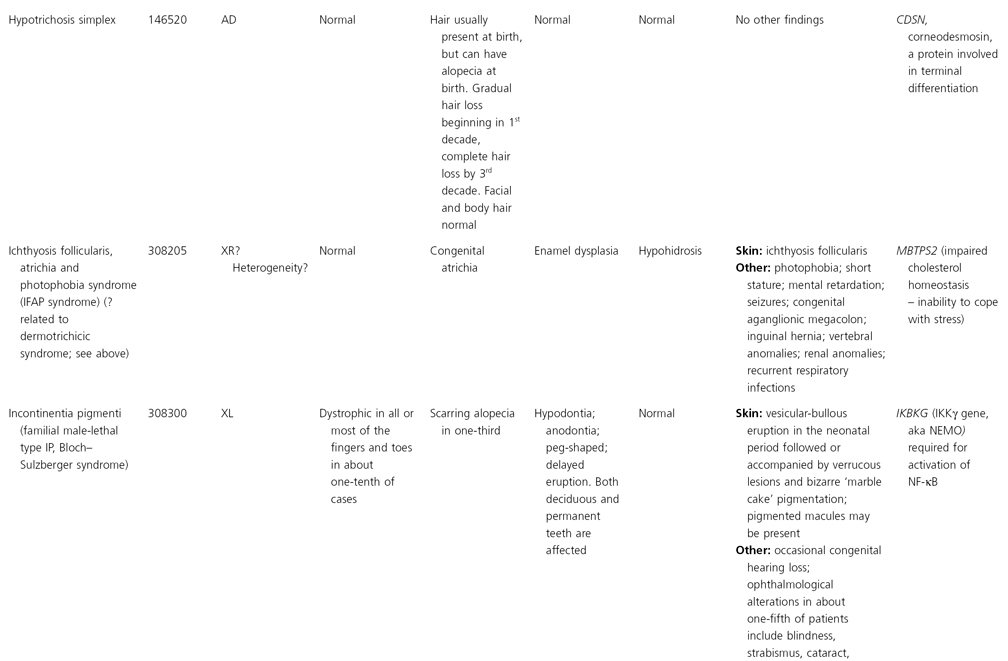

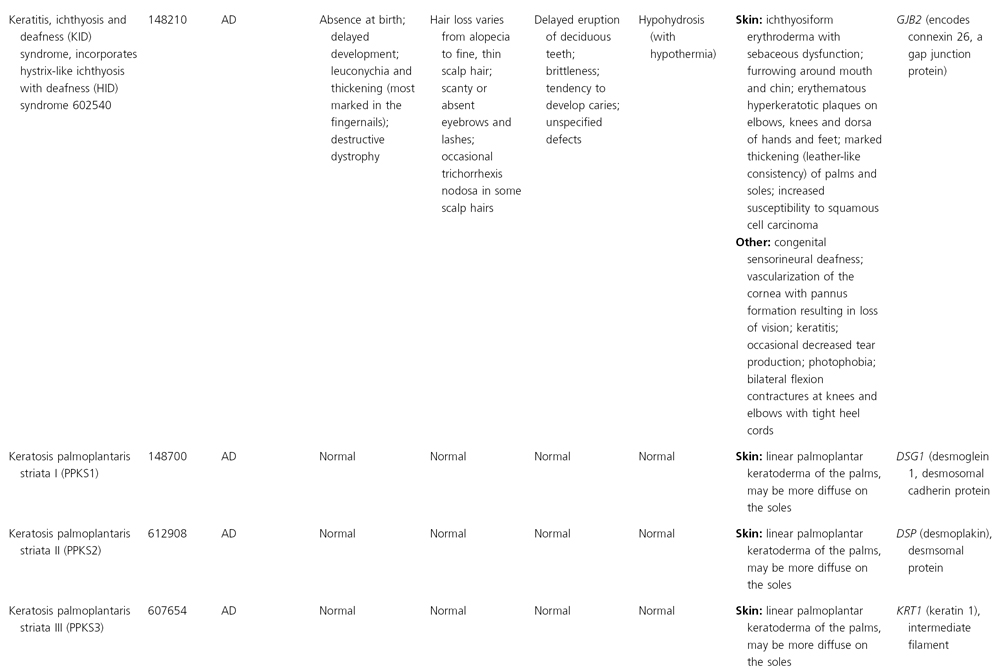
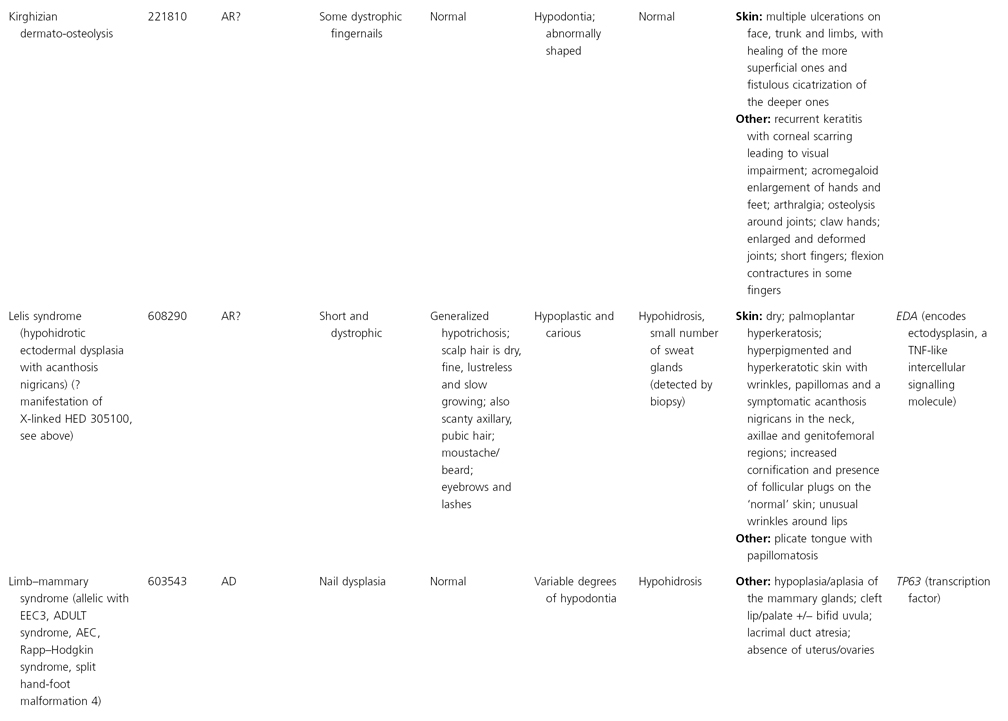
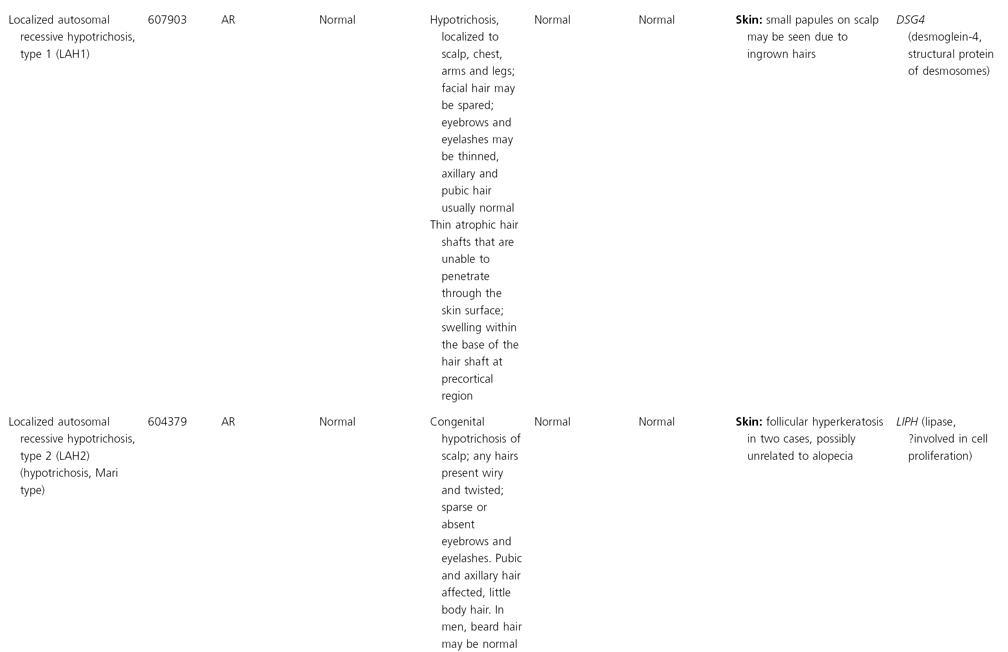
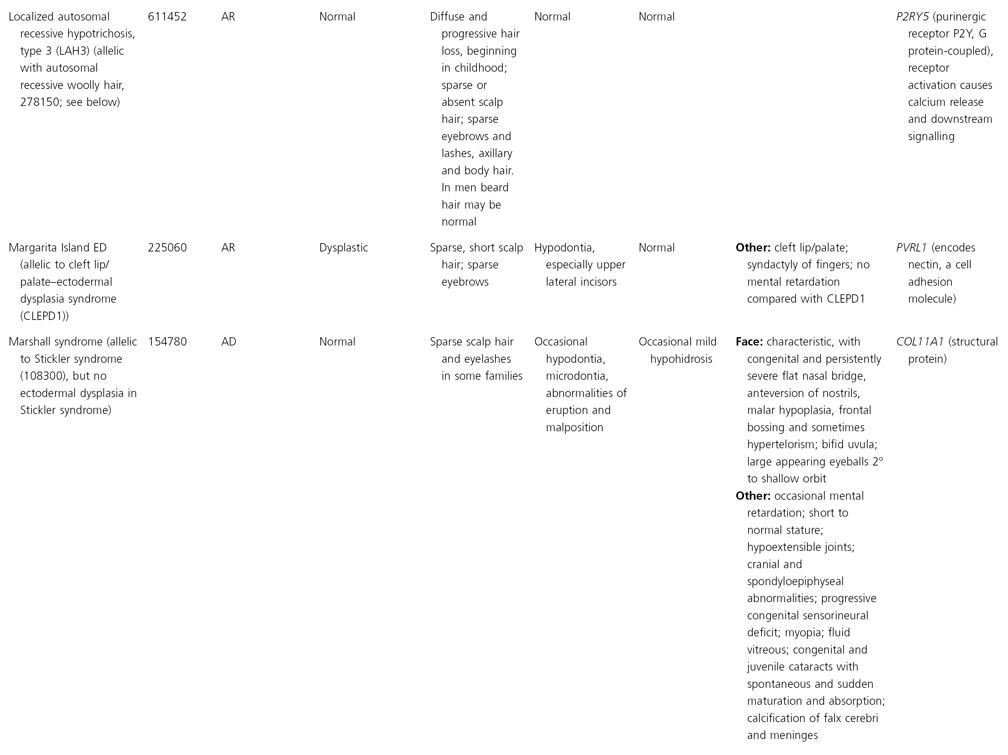
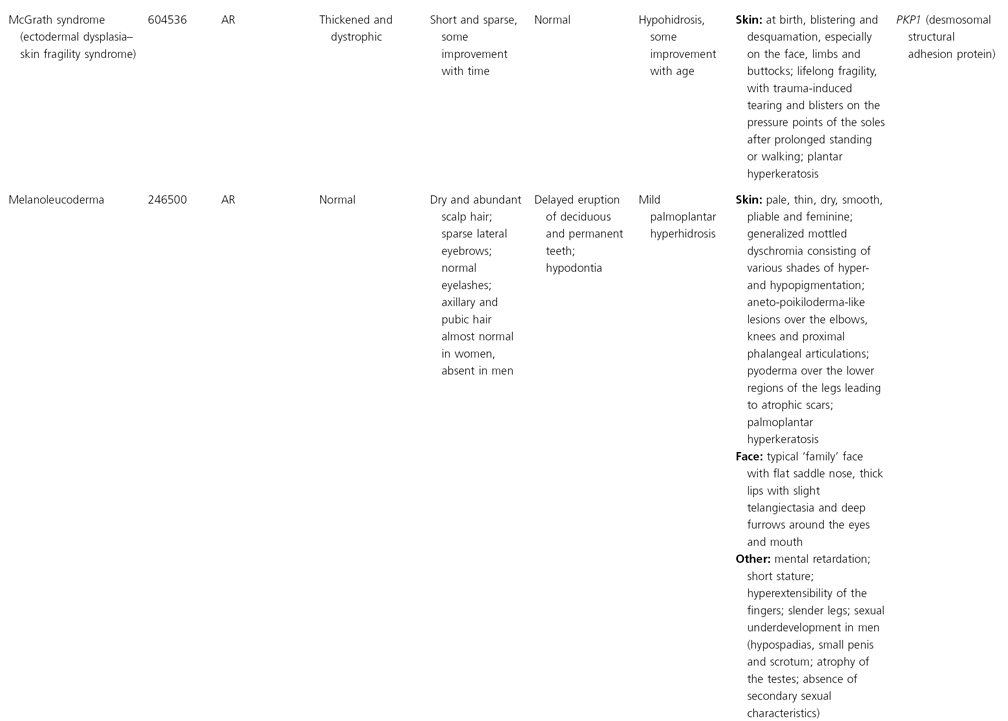
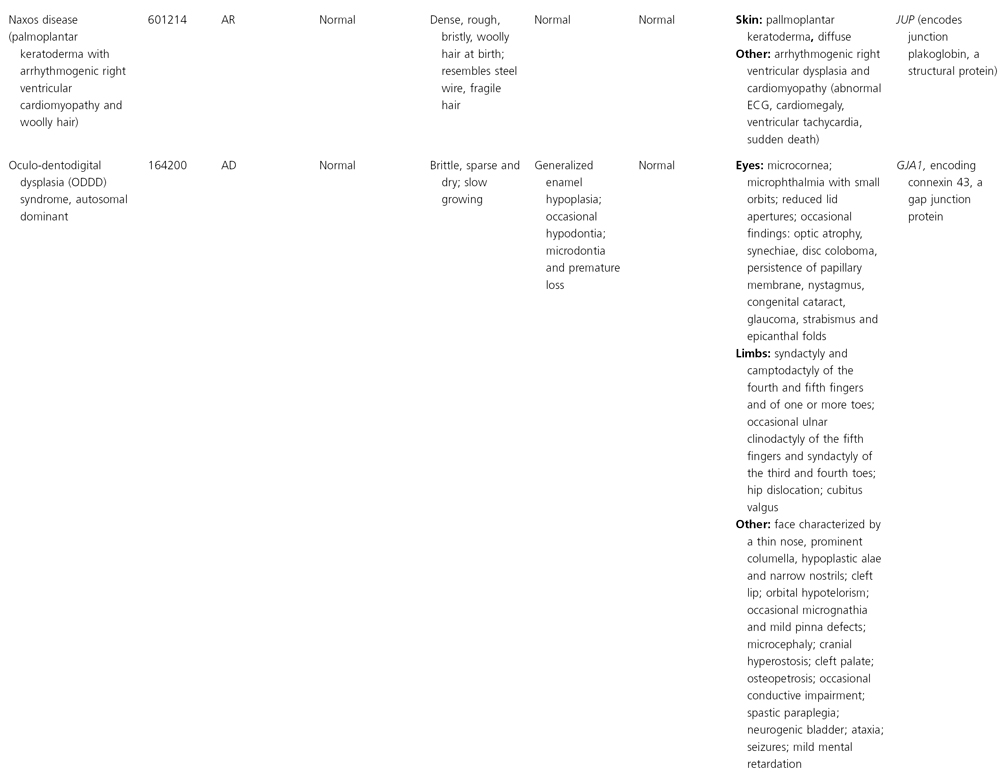
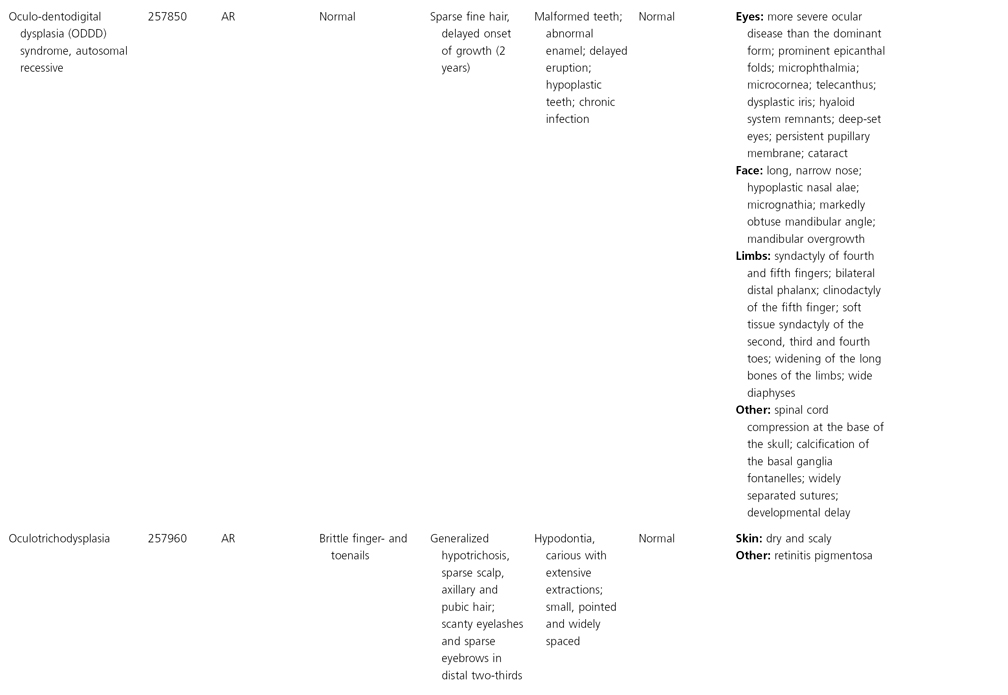

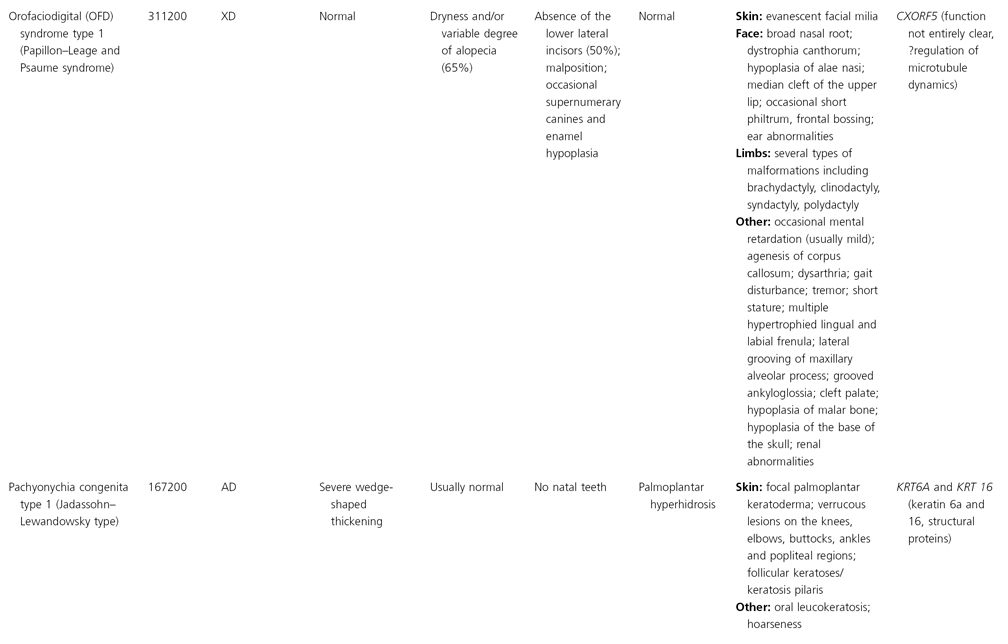
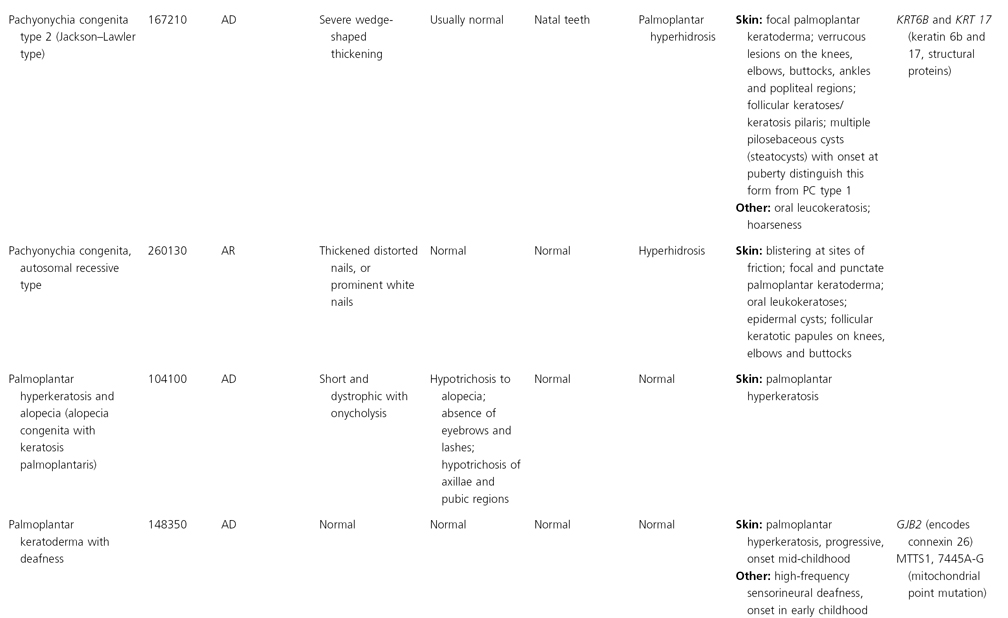
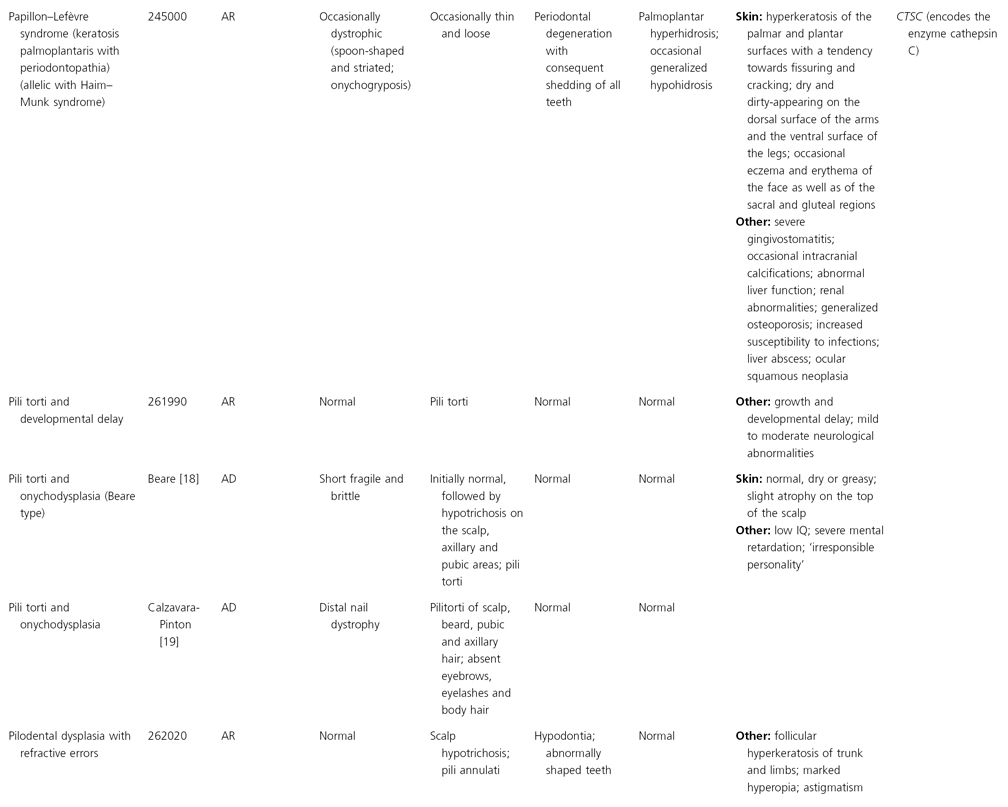
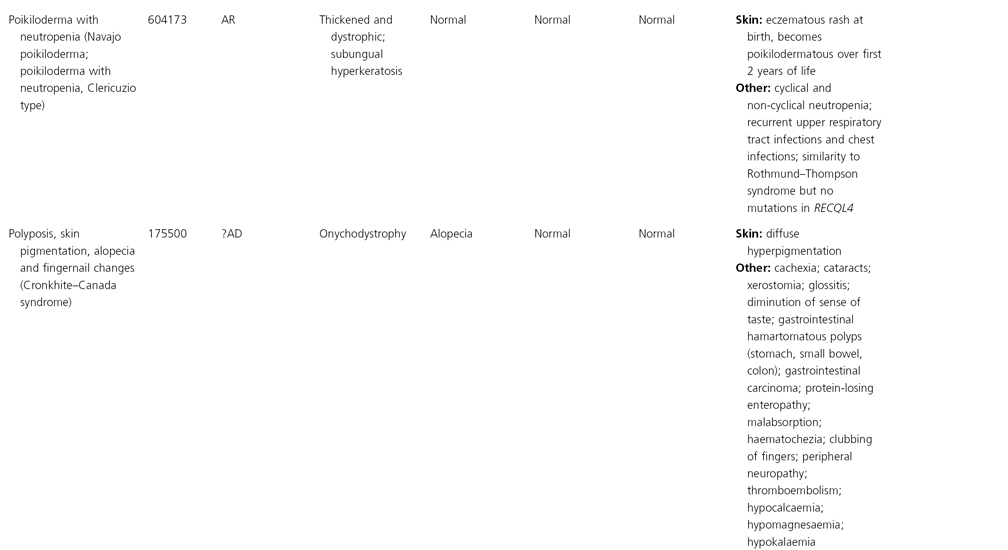

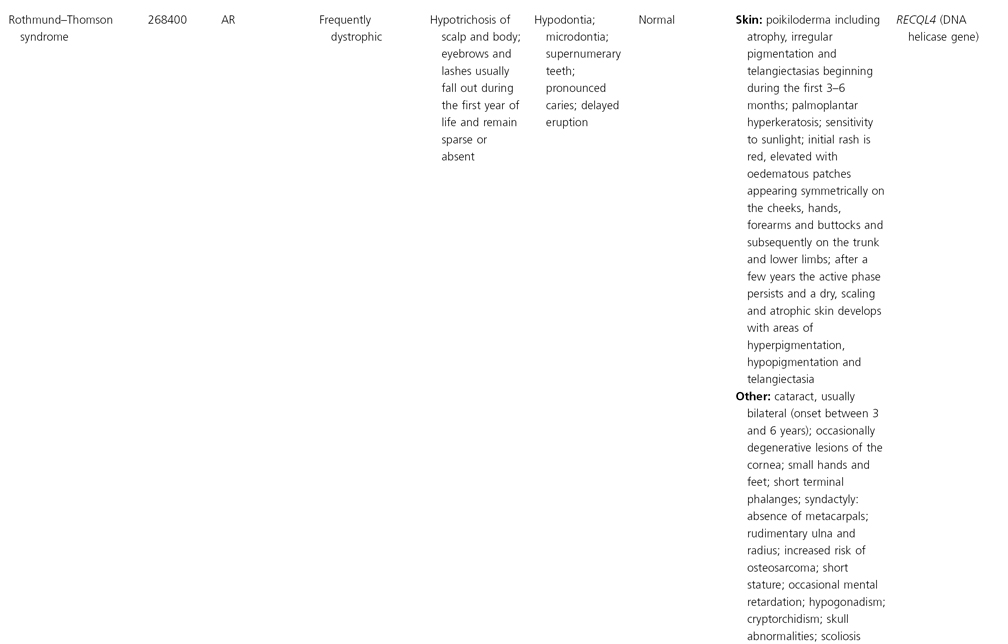
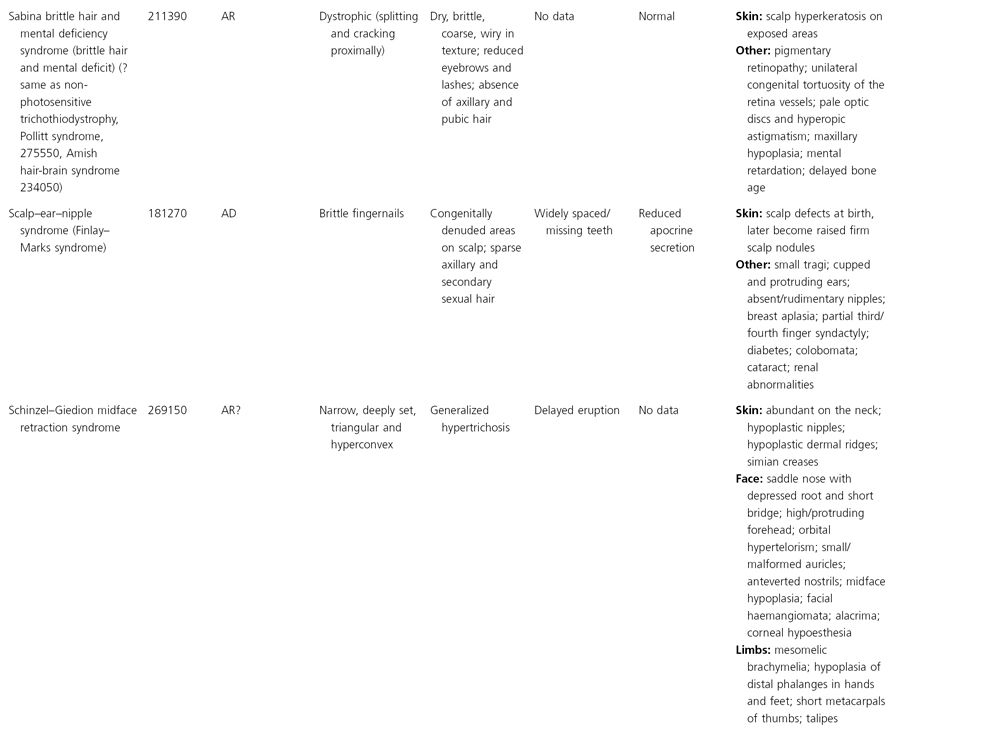

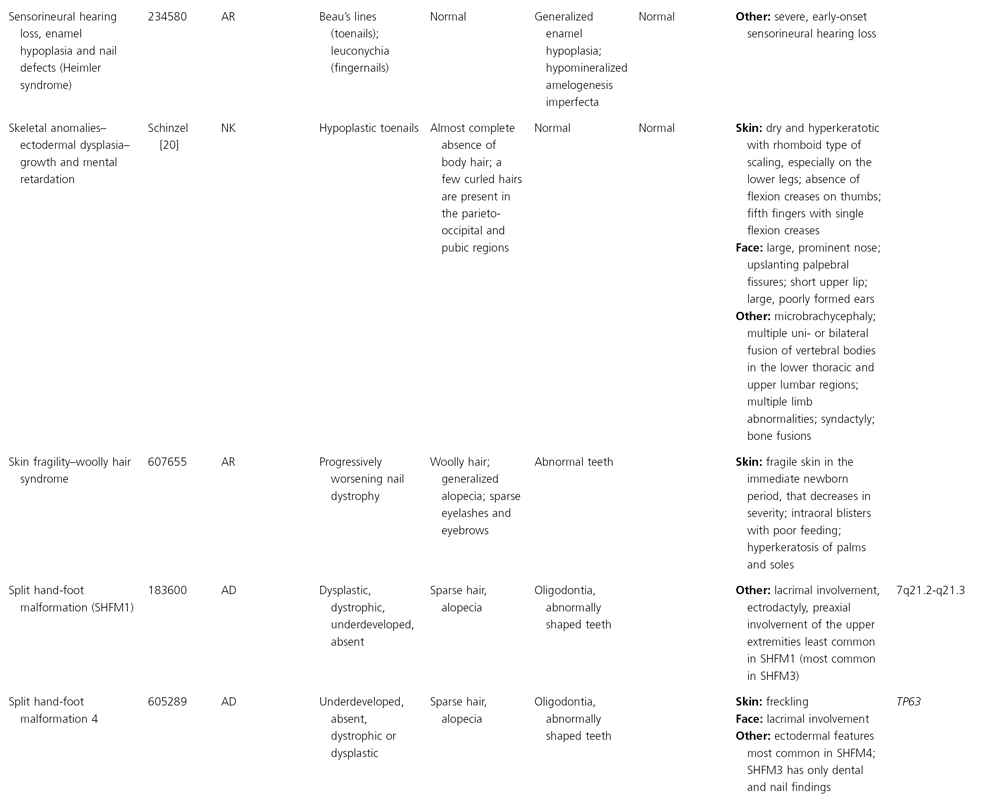
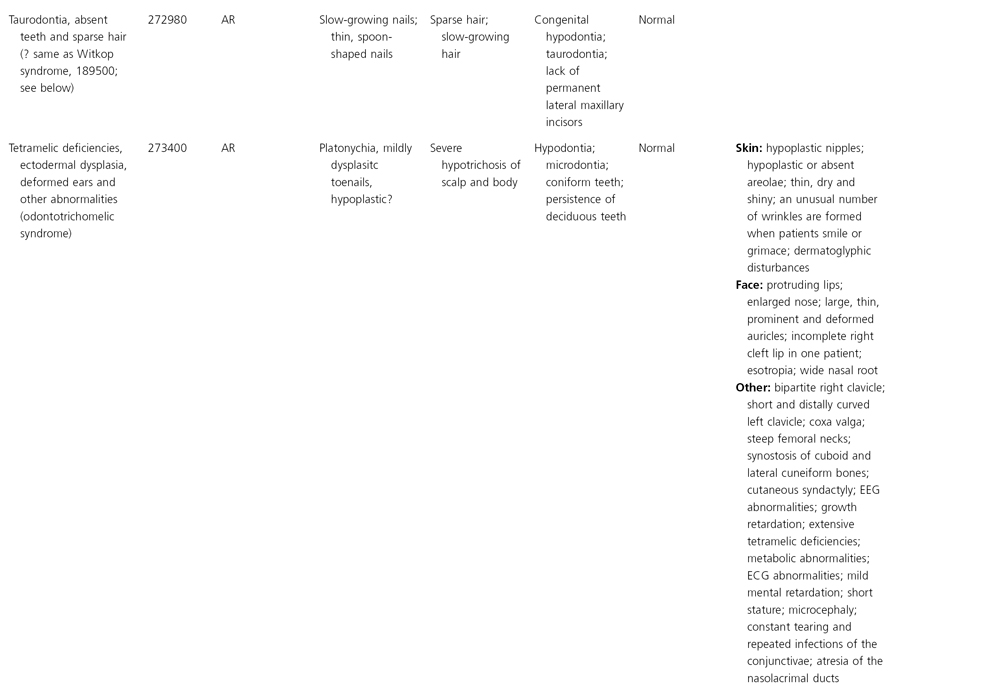
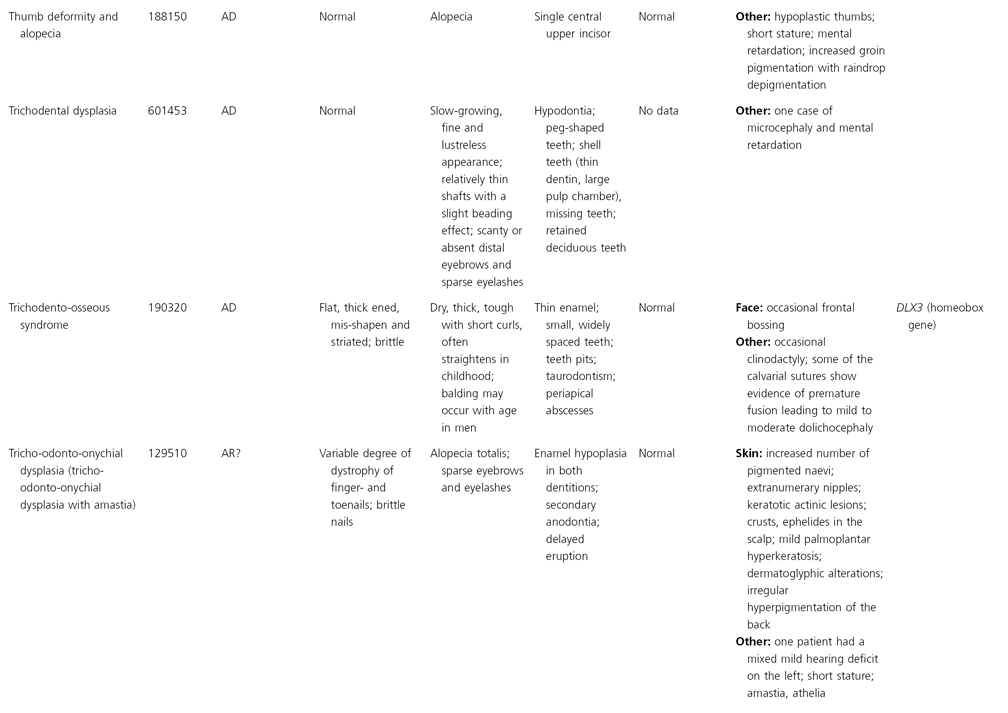

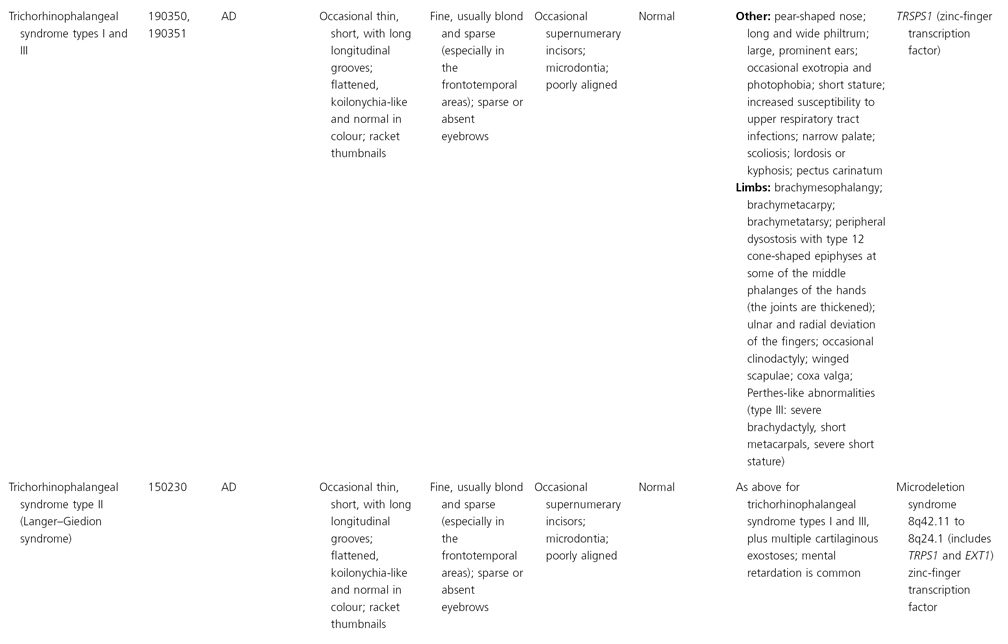
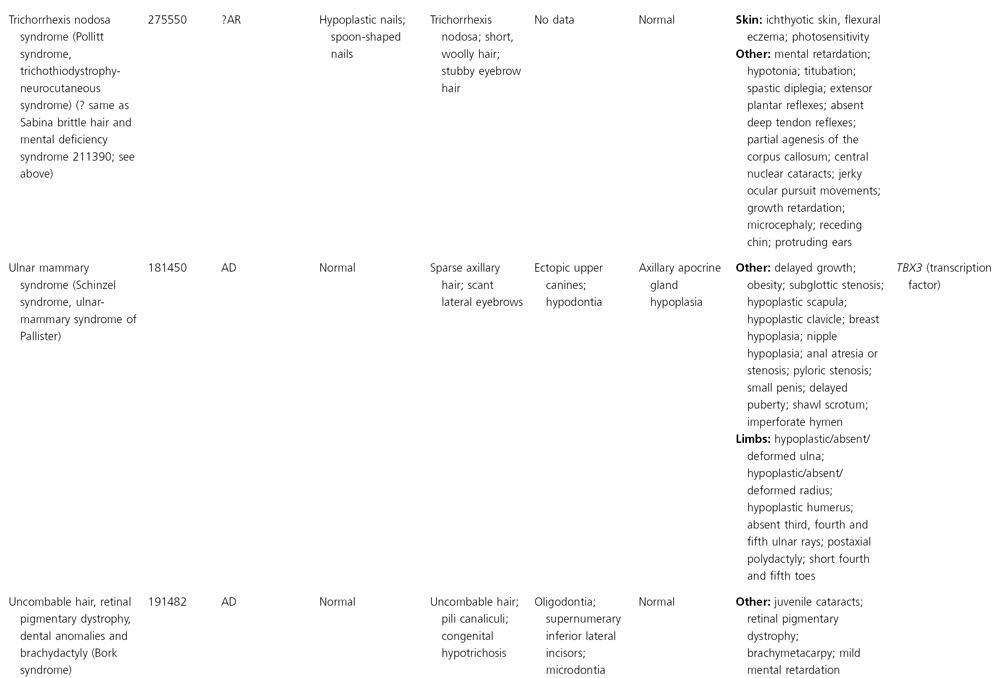
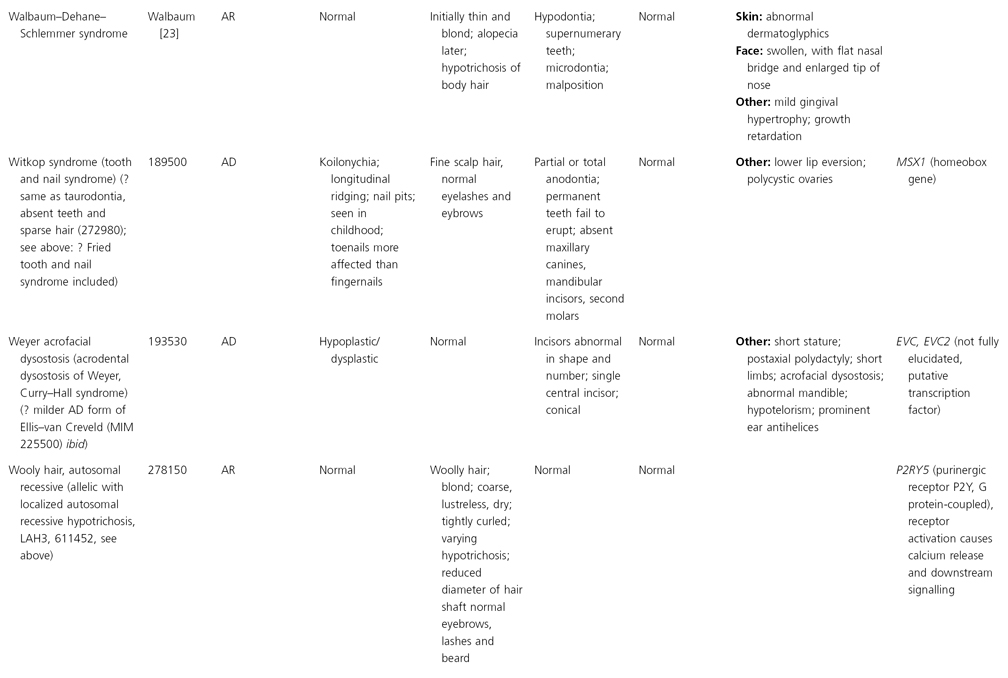
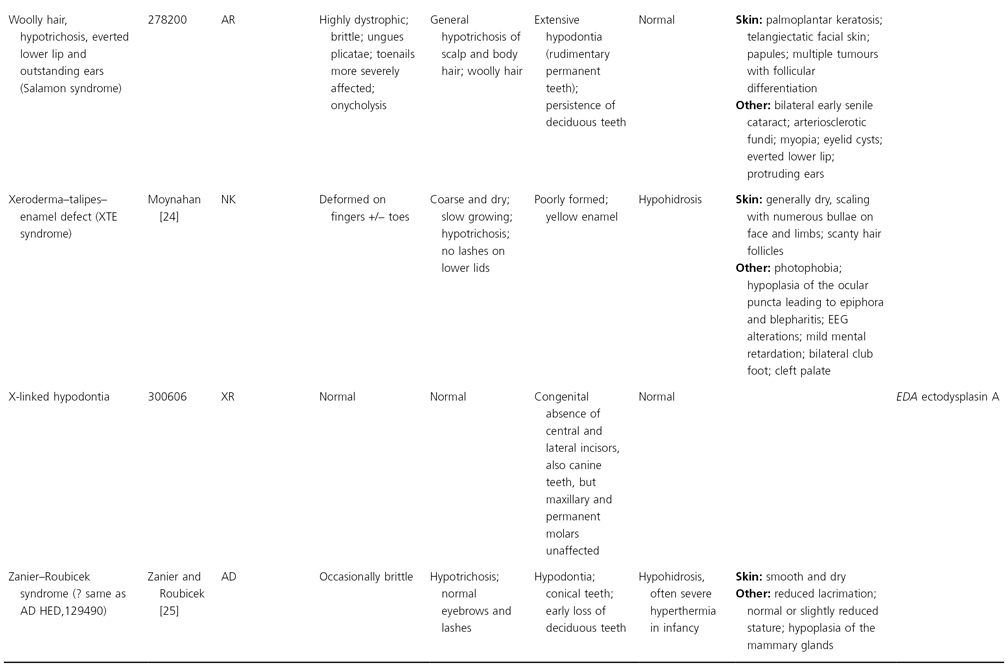
AD, autosomal dominant; AR, autosomal recessive; CGS, contiguous gene syndrome; NK, not known; MIM, Mendelian Inheritance in Man; XL, X-linked.
Ectodermal dysplasias may also be divided into those with isolated involvement of hair, teeth and nails – ‘pure’ EDs – while those with abnormalities of other structures and organs are referred to as ‘ectodermal dysplasia syndromes’. The construction of a practical, convenient classification of ED is made challenging due to the complex interplay between clinical presentation, mode of inheritance and the genes, proteins and molecular pathways involved.
Molecular Insights and New Approaches to Classification
The last decade has seen several important insights into the molecular basis of several of the ectodermal dysplasias. In some cases, the molecular data have confirmed clinical impressions; for example, Hay–Wells syndrome and ectrodactyly–ectodermal dysplasia–clefting (EEC) syndrome have ectodermal dysplasia and clefting of the palate and lip as common clinical features and these conditions are now known to be allelic. In a few conditions, a unifying molecular mechanism has been shown to underlie clinically very distinct conditions. In recognition of the recent advances in understanding the molecular mechanisms underlying these EDs, conditions that share molecular mechanisms are considered together below.
One important concept in ectodermal appendage morphogenesis that recurs throughout the EDs is an early dermal signal initiating morphogenesis, followed by an ectodermal signal to organize the mesenchyme, with a secondary dermal signal to co-ordinate growth and development of the epithelial appendage. For a recent summary, see Fuchs et al. [7]. The molecular mechanisms delineated to date in EDs can be considered under the broad categories of defects in the nuclear factor κB (NF-κB) signalling pathway, p63 transcription factor pathway, Wnt-β-catenin pathway, gap junctions and structural/adhesive molecules. Based on these pathophysiological mechanisms, EDs can also be sorted into two broad categories: Group 1, indicating defects in developmental regulation and epithelial-mesenchymal interaction, and Group 2, indicating defects in proteins of cytoskeleton or adhesion, which are involved in cell–cell communication as well as structural integrity [27,28].
References
1 Online Mendelian Inheritance in Man, OMIM™. McKusick–Nathans Institute for Genetic Medicine, Johns Hopkins University, and National Center for Biotechnology Information, National Library of Medicine, Bethesda, MD. www.ncbi.nlm.gov/omim/.
2 Danz DFG. Sechste Bemerkung. Von Menschen ohne Haare und Zahne. Stark Arch Geburtch Frauenz Neugeb Kinderkr 1792;4:684.
3 Weech A. Hereditary ectodermal dysplasia (congenital ectodermal defect). A report of two cases. Am J Dis Child 1929;37:766–90.
4 Touraine A. L’anidrose hereditaires avec hypotrichose et anodontie (polydysplasie ectodermique héréditaire). Presse Méd 1936;44:145–9.
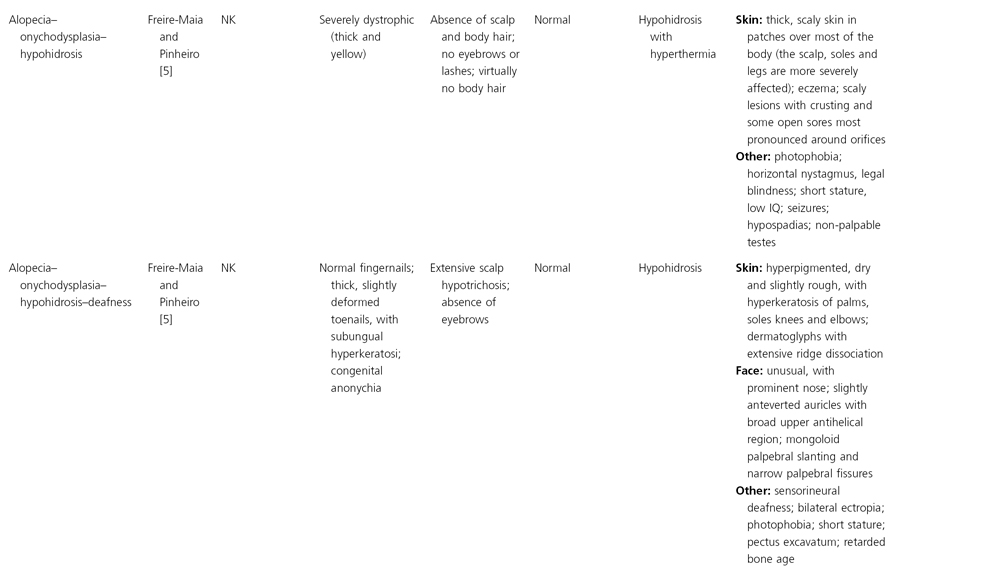
5 Freire-Maia N, Pinheiro M. Ectodermal Dysplasias: a Clinical and Genetic Study. New York: Alan R. Liss, 1984.
6 Pinheiro M, Freire-Maia N. Ectodermal dysplasias: a clinical classification and a causal review. Am J Med Genet 1994;53:153–62.
7 Fuchs E, Merrill BJ, Jamora C et al. At the roots of a never-ending cycle. Dev Cell 2001;1(1):13–25.
8 Lerner AB. Three unusual pigmentary syndromes. Arch Dermatol 1961;83:151–9.
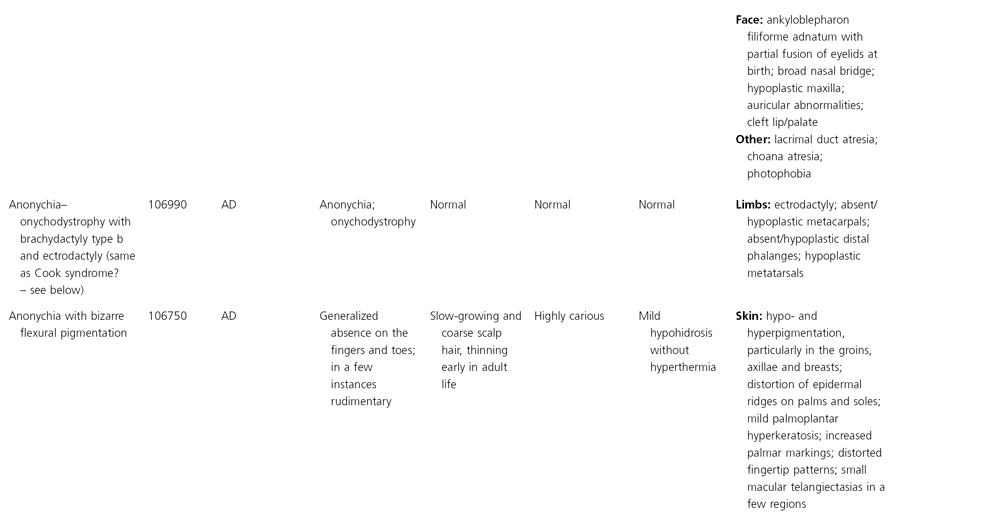
9 Baisch A. Anonychia congenita, Kombiniert mit Polydaktykie and verzogertem abnormen Zahndurchbruch. Dtsch Z Chir 1931;232:450–7.
10 Wallace HJ. Ectodermal defect with skeletal abnormalities. Proc C Soc Med Edinb 1958;51:707–8.
11 Wesser DW, Vistnes LM. Congenital ectodermal dysplasia, anhidrotic, with palatal paralysis and associated chromosome abnormality. Plast Reconstr Surg 1969;8:396–8.
12 Kirman BH. Idiocy and ectodermal dysplasia. Br J Dermatol 1955;67:303–7.
13 Wiedemann HR, Grosse FR, Dibbern H. Caracteristicas das Sindromes em Pediatria. Atlas de Diagnostico Diferencial. São Paulo: Editoria Manole, 1978.
14 Fischer H. Familiar hereditares Vorkommen von Keratoma palamare et plantare, Nagelverandergungen, Haaranomalien und Verdickung der Endglieder der Finger und Zehen in 5 Generationen (die Beziehungen dieser Veranderungen zur inneren Sekretion). Dermatol Zeitschr 1921;32:114–42.
15 Fried K. Autosomal recessive hydrotic ectodermal dysplasia. J Med Genet 1977;14:137–9.
16 Jorgenson RJ. Gingival fibromatosis. Birth Defects 1971;VII:278–80.
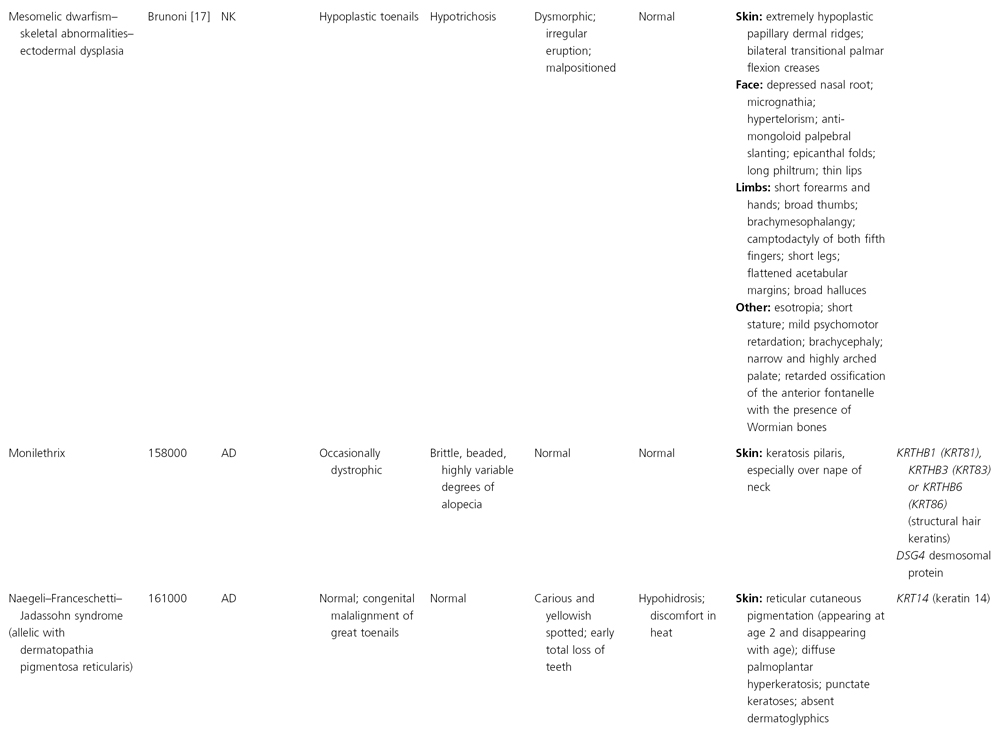
17 Brunoni D, Lederman H, Ferrari S et al. Uma sindrome malformativa com nanismo mesomelico, malformacoes esqueleticas, displasia ectodermica e facies tipica. Cienc Cult 1982;34:694.
18 Beare JM. Congenital pilar defect showing features of pili torti. Br J Dermatol 1952;64:366–72.
19 Calzavara-Pinton P, Carlino A, Benetti A et al. Pili torti and onychodysplasia. Report of a previously undescribed hidrotic ectodermal dysplasia. Dermatologica 1991;182:184–7.
20 Schinzel A. A case of multiple skeletal anomalies, ectodermal dysplasia, and severe growth and mental retardation. Helv Paediatr Acta 1980;35:243–51.
21 Pinheiro M, Pereira LC, Freire-Maia N. A previously undescribed condition: tricho-odonto-onycho-dermal syndrome. A review of the tricho-odonto-onychial subgroup of ectodermal dysplasias. Br J Dermatol 1981;105:371–82.
22 Koshiba H, Kimura O, Nakata M et al. Clinical, genetic, and histologic features of the trichoonychodental (TOD) syndrome. Oral Surg Oral Med Oral Pathol 1978;46:376–85.
23 Walbaum R, Dehaene P, Schlemmer H. Dysplasie ectodermique: une forme autosomique recessive? Arch Fr Pediatr 1971;28:435–42.
24 Moynahan EJ. XTE syndrome (xeroderma, talipes and enamel defect): a new heredo-familial syndrome. Proc Roy Soc Med Lond 1970;63:1–2.
25 Zanier JM, Roubicek MM. Hypohidrotic ectodermal dysplasia with autosomal dominant transmission. Fifth International Congress on Human Genetics, Mexico, 1976: Communication 273.
26 Priolo, M, Laganà C. Ectodermal dysplasias: a new clinical-genetic classification. J Med Genet 2001;38:579–85.
27 Priolo M. Ectodermal dysplasias: an overview and update of clinical and molecular-functional mechanisms. Am J Med Genet A 2009;149A(9):2003–13.
Ectodermal Dysplasias Due to Mutations in Tumour Necrosis Factor-Like/NF-κB Signalling Pathways
Overview of Molecular Pathways
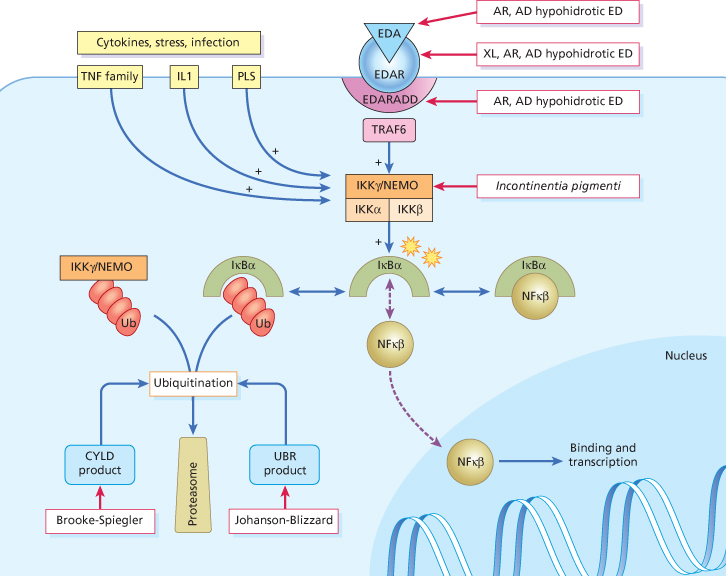
The transcription factor NF-κB regulates the expression of multiple genes with functions in controlling the immune and stress responses, cell adhesion, protection against apoptosis and inflammatory reactions [1,2]. NF-κB is composed of homo- or heterodimers of five proteins belonging to the Rel family (p50, p52, c-rel, relA and relB). NF-κB is usually maintained in an inactive state within the cytoplasm by association with inhibitory proteins of the IB (IκB) family: IBα, IBβ and IBε. IB molecules are phosphorylated on two critical serine residues in response to multiple stimuli such as cytokines, various stress signals and viral and bacterial infections. An increasing number of signals that initiate this phosphorylation are identified year-on-year, but the best studied signals include the tumour necrosis factors (TNFs), lipopolysaccharides (LPS) and interleukin 1 (IL-1). Phosphorylation at these sites allows IB molecules to be recognized by a ubiquitination complex; following polyubiquitation, IBs are degraded by proteasomes, thus freeing free NF-κB to enter the nucleus and activate target genes [3].
The kinase that phosphorylates IB has been designated IKK (for IκB kinase) and has been shown to consist of two catalytic subunits (IKKα/IKK1 and IKKβ/IKK2) and a third component IKKγ (more commonly known as NEMO – NF-κB essential modulator) that provides a structural and regulatory function to the complex. Cell lines lacking NEMO are unable to activate NF-κB in response to most stimuli [4]. Extensive work with mouse models has confirmed the centrality of the NF-κB pathway in apoptosis and inflammatory and immune functions [5]. Complete absence of NF-κB leads to prenatal death owing to massive TNF-induced liver apoptosis, and more subtle knockouts that alter NF-κB activity all lead to immune defects. IKKα is also an important suppressor of skin cancer [6].
The elucidation of the NF-κB pathway has recently generated much interest in the EDs, and defects at various levels have been identified in several EDs. In many cases, these predominantly phenotype-driven, mouse–human comparison studies have yielded significant new insights in molecular pathways (Table 127.2) [1]. One of the best characterized pathways in NF-κB activation is the ectodysplasin pathway, an upstream activator (Fig. 127.1). Since 1997, defects in this pathway have been demonstrated in the X-linked, autosomal dominant and recessive subtypes of hypohidrotic ED (HED). Subsequently, mutations in downstream components have been shown to underlie familial incontinentia pigmenti and HED associated with immunodeficiency and/or osteopetrosis and HED associated with T-cell immune deficiency.
Table 127.2 Genotype–phenotype correlation in disorders due to mutations in the NF-κB signalling pathway
| Gene | Mouse phenotype | Human disease (MIM number; abbreviation) |
| EDAR | Downless | Autosomal dominant hypohidrotic ectodermal dysplasia (129490) and autosomal recessive hypohidrotic ectodermal dysplasia recessive (224900) |
| EDA1 | Tabby | X-linked hypohidrotic ectodermal dysplasia (305100) |
| EDARADD | Crinkled | Autosomal recessive hypohidrotic ectodermal dysplasia (606603) and autosomal dominant hypohidrotic ectodermal dysplasia (129490) |
| TRAF6 | Hypohidrotic ED (in knockout mice), abnormal teeth, osteopetrosis | Familial osteoporosis (611739) |
| NEMO/IKKγ | Heterozygotes for NEMO deficiency develop: unique dermatopathy with keratinocyte hyperproliferation, skin inflammation, hyperkeratosis and increased apoptosis. +/− females recover, −/− males die in utero | Incontentia pigmenti (308300; IP) |
| NEMO/IKKγ (hypomorphic mutations) | As above | Hypohidrotic ectodermal dysplasia with immune deficiency (300291; ED-ID) |
| NEMO/IKKγ (stop codons) | As above | Anhidrotic ectodermal dysplasia with immunodeficiency, osteopetrosis and lymphoedema (300248;OL-EDA-ID) |
| IkBα (hypermorphic dominant mutations) | Not known | Autosomal dominant ectodermal dysplasia with a severe and unique T-cell immunodeficiency (612132; EDA-ID) |
| IRAK (amorphic mutations) | Severe impairment in interferon-γ production and the induction of natural killer cell cytotoxicity by IL-18 | None yet identified; ? lupus susceptibility |
Fig. 127.1 Schematic of NF-κB pathway. EDA binds to EDAR, which interacts with EDARADD. Downstream signalling from this complex, as well as input from cytokines and other stressors, leads to activation of the IKK (inhibitor κ B kinase) complex via TRAF 6 and other signalling molecules. When activated, the IKK complex, which is made up of three subunits (α, β and γ), phosphorylates IκB α (inhibitor κ B α), which results in its targeting for ubiquitination and degradation. This causes the release of NF-κB, allowing it to enter the nucleus, bind to transcriptional elements, and cause upregulation of NF-κB mediated gene targets. ED, ectodermal dysplasia; AD, autosomal dominant; AR, autosomal recessive; XL, X-linked; LPS, lipopolysaccharides; Ub, ubiquitin; EDAR, ectodysplasin A; EDARADD, EDAR-associated death domain.
The X-linked HED gene, EDA, which maps to Xq12–13.1 and is also mutated in the mouse orthologue tabby [7], was first described in 1996 [8]. Canine and bovine models for X-linked HED have mutations in the similar genes [9–11] EDA encodes two isoforms of a transmembrane protein, ectodysplasin-A (EDA), that has homology to the TNF family. The extracellular domain of EDA has a collagen-like repeat and a furin cleavage site, unique in TNF proteins. Cleavage is necessary to enable solubility and functionality of EDA. The two longest isoforms, EDA-A1 and EDA-A2, bind to two different receptors: EDA-A1 binds to the EDAR protein and EDA-A2 binds to another X-linked receptor, XEDAR [12]. Mutations have been identified in all domains of EDA in patients with HED; many of these mutations are thought to have an effect on solubility or cleavage of ectodysplasin-A, rendering it non-functional [13]. Mutations in the EDA Gly-X-Y domain are thought to prevent bundling of EDAR trimers but do not appear to have an effect on EDA–EDAR binding (B. Ferguson, personal communication, 2003). EDA mutations are also responsible for X-linked hypodontia (MIM 300606), a condition notable for the congenital absence of incisors but not molars [14].
The physiological role of EDA in hair follicle morphogenesis was reinforced by the isolation of the gene for autosomal dominant/recessive HED. Patients with autosomal dominant or recessive HED are phenotypically identical to those with X-linked HED. The mutated gene, previously named downless (DL) after the mouse homologue, encodes a member of the tumour necrosis factor receptor (TNFR) superfamily which functions as an ectodysplasin receptor (EDAR) [15]. Loss-of-function mutations throughout EDAR have been reported in autosomal recessive HED and dominant negative mutations have been reported in autosomal dominant HED within the death domain of this transmembrane protein [16].
The EDA–EDAR pathway was further refined when the molecular basis of a third mouse homologue was identified. The crinkled mouse (cr) is a spontaneous mouse mutant with an identical phenotype to downless and tabby. Using positional cloning techniques, the causative gene was identified in an adapter protein (EDAR-associated death domain, termed Edaradd) for the EDA–EDAR complex [17]. The same group also identified mutations in a family with autosomal recessive HED [17]. The Edaradd death domain interacts with the intracellular death domain of EDAR, linking it to downstream signals leading to NF-κB activation [17]. Edaradd associates with TNFR-associated factor (TRAF) 1, 2 and 3. The gene encoding the Edaradd protein, EDARRADD, may be mutated in both autosomal recessive and dominant hypohidrotic ectodermal dysplasia [18,19].
NF-κB activation by the EDAR pathway is NEMO dependent [20], and the relevance of this interaction to human ectodermal dysplasias became clear when loss-of-function mutations were identified in the IKKγ gene (NEMO) in incontinentia pigmenti [21]. This discovery was followed by identification of less critical mutations in NEMO in several male patients with an unusual phenotype of HED associated with immune deficiency (EDA-ID) [20,22]. Mutations in the coding region are associated with the EDA-ID phenotype, and specific mutations in the stop codon of NEMO cause a more severe syndrome of osteoporosis and/or lymphoedema associated with EDA-ID [20].
Recently, two other EDAR-related members of the TNFR superfamily, X-linked ectodysplasin-A2 receptor (XEDAR) [12] and TNFR superfamily member 19 (TNFRSF19), also known as TROY or TAJ (toxicity and JNK inducer) [23,24], have been reported. Signals from each of these receptors were shown to activate NF-κB, providing further candidate genes and candidate signalling systems for human HED. TRAF-6 is a cytoplasmic adapter protein that links signals from members of the TNFR superfamily to activation of transcription factors such as NF-κB through IKK activation. TRAF-6 −/− mice display HED, revealing yet more complexity to these signalling systems [25]. It is likely that several of these genes will, in time, be shown to have relevance in human HED.
Regulatory modulators of the NF-κB system may also prove relevant to ectodermal dysplasia in the future; recently mutations in a gene encoding a protein involved in deubiquitinization of inhibitor proteins has been found in familial cylindromatosis (Brooke–Spiegler syndrome, MIM 605041), and its allelic disorders, familial cylindromatosis (MIM 132700) and multiple familial trichoepitheliomas (MIM 601606). Mutations in the cylindromatosis gene (CYLD) are associated with these disorders, and this protein has an important role in negative regulation of the NF-κB pathway, as it deubiquitinates multiple NF-κB regulators, including TRAF2, TRAF6 and NEMO [26–30]. Johanson–Blizzard syndrome, another ectodermal dysplasia, is due to mutations in UBR1, which encodes ubiquitin-protein ligase E3 component N-recognin, a protein that binds to a destabilizing N-terminal residue of a substrate protein and participates in the formation of a substrate-linked multiubiquitin chain [31].
X-Linked, Autosomal Dominant and Recessive Hypohidrotic Ectodermal Dysplasia
The phenotypic appearances of the X-linked (MIM 305100), autosomal recessive (MIM 606603) and autosomal dominant types (MIM 129490) are identical.
Definition.
X-linked HED is the most common of the ectodermal dysplasias and is characterized by hypotrichosis, hypodontia, hypohidrosis and distinctive facial features. Autosomal recessive HED is clinically identical to X-linked HED and females are as severely affected as males.
History.
X-linked HED was first described in 1848 by Thurnam [32]. In 1921, Thadani [33] determined that it was an X-linked disorder and later reported that female carriers manifest varying signs of the condition.
Pathology.
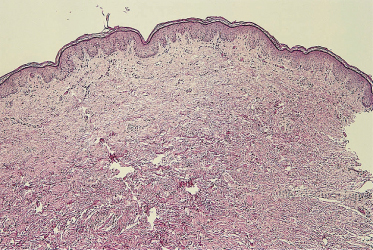
The epidermis is thin, with effacement of rete ridges. The striking finding is absent or sparse eccrine glands and ducts in affected males (Fig. 127.2) [34,35]. Hair follicles and sebaceous glands are variably reduced in number and may appear rudimentary [34–36]. Apocrine glands may be absent, sparse or even normal. The nasal mucosa demonstrates almost complete loss of ciliated cells [37]. Mucous glands of the upper respiratory tract may be sparse or absent [34]. Mucus-secreting glands in the duodenum may also be absent [35]. Light and scanning electron microscopic findings of hair shaft abnormalities are variable and include longitudinal clefts or grooves and transverse fissuring. The bulb of the hair shaft is dystrophic in some individuals [38]. Radiographs of the mandible reveal dental hypoplasia or aplasia [34,39].
Fig. 127.2 Skin biopsy from the trunk of an affected male with X-linked HED. Note the absence of hair follicles, sebaceous glands and eccrine glands (haematoxylin and eosin; 10× original magnification).
Clinical Features
Hair
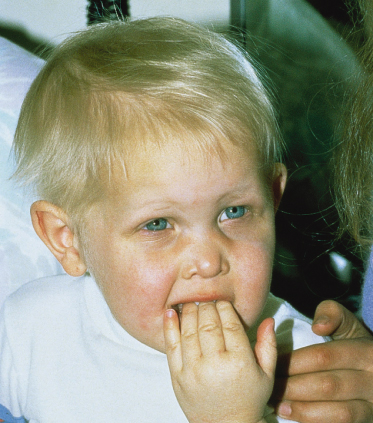
The scalp hair is sparse, fine and lightly pigmented, and grows slowly (Fig. 127.3). Eyebrows are scanty or absent; sometimes just the outer two-thirds are missing. The eyelashes may be normal, sparse or completely absent. Secondary sexual hair in the beard, pubic and axillary regions is variably present and may be normal. Hair on the torso and extremities is usually absent [34,36,40,41]. Approximately 70% of obligate female carriers of X-linked HED describe their hair as being sparse or fine [40].
Fig. 127.3 Young boy with X-linked HED. The hair is fine, lightly pigmented and sparse.
Courtesy of Dr Virginia Sybert.
Teeth
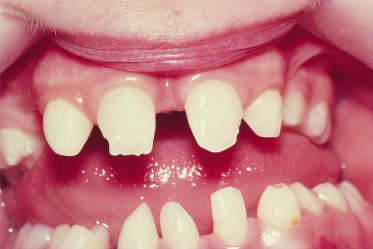
Dental abnormalities vary from complete absence of teeth to sparse, abnormally shaped teeth. Studies reveal a mean of 24 missing teeth, out of a total of 28, in affected males [40,42]. Dentition is delayed and the erupted teeth tend to be small and widely spaced, and are frequently conical or peg-shaped. Both deciduous and permanent teeth are affected. The alveolar ridges are hypoplastic (Fig. 127.4), which gives rise to full, everted lips [39,43]. About 80% of obligate female carriers of X-linked HED have distinct dental abnormalities, including absent permanent teeth and small or peg-shaped teeth (Fig. 127.5) [40]. Oligodontia in the primary dentition is an important clinical predictor of EDA mutation in females [44].
Fig. 127.4 Hypoplastic aveolar ridges and peg-shaped teeth in an affected male with X-linked HED.
Courtesy of Dr Virginia Sybert.
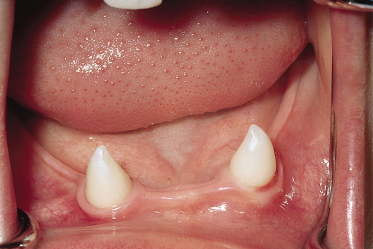
Fig. 127.5 This female heterozygous for X-linked HED exhibits abnormally shaped and absent permanent teeth.
Courtesy of Dr Virginia Sybert.
Nails
The nails are normal in most individuals. Thin, brittle nail plates with longitudinal ridges have been described in some individuals.
Sweat Glands
Sweating is severely diminished or absent owing to a paucity or absence of eccrine glands. The ability to thermoregulate by evaporative cooling is inadequate and hyperthermia can occur with physical exertion or in a warm environment. This is particularly problematic in infants and young children, who may experience recurrent bouts of fever as high as 42°C. Heat intolerance does occur in older children and adults, but they learn to control their body temperature by drinking cold liquids, wetting their skin or clothing and seeking out cool surroundings [40]. About 25% of heterozygote females experience heat intolerance, and almost half notice that their ability to sweat is reduced [40]. The hypohidrotic areas of skin in carrier females of X-linked HED occur in defined linear patterns corresponding to the lines of Blaschko [45]. Diminished apocrine sweating in affected individuals is not problematic.
Skin
At birth, affected males may demonstrate marked scaling or peeling of their skin that may be mistaken for a collodion membrane [46]. In children and adults, the skin is fine, smooth and dry. Periorbital hyperpigmentation and fine wrinkling around the eyes are characteristic features of the disorder. Eczema is common and is prominent in flexural areas [41,47]. Small milia-like papules may be found on the face [36,48].
Other Ectodermal Structures
Diminished or absent salivary glands and mucous glands of the nose, mouth and ears cause numerous otolaryngological complications including nasal obstruction caused by thick, foetid nasal discharge and adherent nasal crusts, sinusitis, recurrent upper respiratory tract infections, feeding problems in infancy, xerostomia, hoarse voice and impacted cerumen [36,47–49]. Diminished production of tear film from the lacrimal glands may cause dry eyes, photophobia and corneal damage [48,50]. A third of affected males have abnormalities of the nipples including absent, simple or accessory nipples [40,51]. Female carriers may also be affected with marked breast asymmetry, inadequate breast milk production or athelia. Pituitary and adrenal insufficiency have been reported [48].
Craniofacial Features
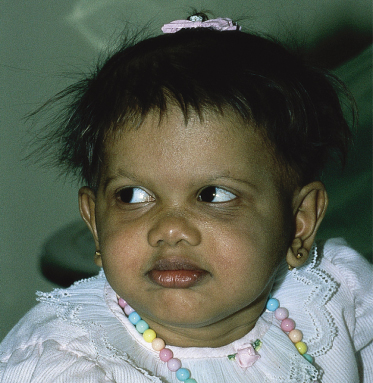
The facies are distinctive with relative frontal bossing, concave midface, saddle nose and everted lips [34,36,39]. A third of affected males have ears that are described as simple or satyr [40]. The distinctive facial features may not be obvious at birth but become more noticeable with age (Figs 127.6, 127.7).
Fig. 127.6 The facial features of X-linked HED may not be obvious at birth, but become more pronounced over time.
Courtesy of Dr Virginia Sybert.
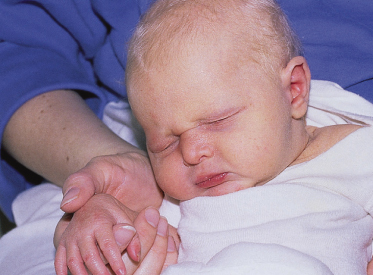
Fig. 127.7 This female exhibits a concave midface, saddle nose and everted lips.
Courtesy of Dr Virginia Sybert.
Other Clinical Features
Diminished or absent mucous glands of the tracheal, bronchial, oesophageal, gastric and colonic mucosa cause problems with recurrent bronchitis, pneumonia, dysphagia and gastro-oesophageal reflux and constipation [40,46,48]. Reactive airways associated with wheezing are a common problem [40]. There is no convincing evidence for thyroid or parathyroid abnormalities, nor for a primary immune deficiency associated with X-linked HED [40].
Prognosis.
Failure to thrive occurs in up to 40% of affected males [40]. Height and weight are compromised in early childhood but appear to normalize with time. Mortality in infancy and early childhood is historically 25%, primarily owing to hyperthermia, failure to thrive and respiratory infections [40]. Febrile seizures can occur with hyperpyrexia [40,47]. Speech problems may exist as a result of hypodontia, nasal obstruction and impacted cerumen [47,49].
Differential Diagnosis.
Affected infants with scaling skin may be misdiagnosed as collodion babies with lamellar ichthyosis. The saddle nose and abnormal teeth have caused diagnostic confusion with congenital syphilis [36,48]. Once the characteristic facies and lack of sweating are evident, there are very few disorders to consider in the differential diagnosis. HED with hypothyroidism displays hypohidrosis with hyperthermia and hypotrichosis but the teeth are normal, the nails are significantly dystrophic and the skin has mottled-brown areas of pigmentation [52]. Fried tooth and nail syndrome manifests as hypotrichosis, hypodontia and prominent everted lips, but sweating is normal [52]. Basan syndrome is characterized by hypotrichosis, hypodontia and hypohidrosis but also by severe nail dystrophy and congenital absence of dermatoglyphics [40].
For the purposes of genetic counselling and reproductive planning, it is possible to perform DNA-based molecular genetic diagnosis in selected patients. This can help to conclusively distinguish between X-linked and autosomal forms where there is no family history to indicate the mode of inheritance.
Treatment.
A multidisciplinary approach to the management of these individuals is advocated [52]. Early diagnosis is crucial to avoid life-threatening complications in infancy, for planning long-term management and to define recurrent risks for families [53]. Female carriers may be detected in most cases by careful clinical examination for patchy distribution of scalp and body hair, sweat pores and hypodontia [47]. DNA-based molecular diagnosis in affected families can detect female carriers of X-linked HED with considerable accuracy. DNA-based prenatal diagnosis is also possible in families at risk for the disorder.
Prevention of hyperthermia is critical. This is done by avoiding heat and physical overexertion, cooling the body with wet clothing and cool drinks and by air-conditioning home and school environments. Early dental restoration with bonding, overdentures or implants is imperative [49,54,55]. Nasal crusting and discharge can be managed with saline nose drops and a home humidifier. Consumption of large amounts of liquids or the use of artificial saliva preparations minimizes dry mouth and swallowing difficulties [56]. Dry eyes may be treated with artificial tears. The daily use of lubricating drops facilitates the removal of impacted earwax. Pulmonary difficulties are managed by avoidance of smoky, dusty environments, adequate humidification and the use of chest physiotherapy and antibiotics when appropriate [57].
Studies on animal models of X-linked HED suggest the possibility of more targeted therapies for this condition in the future, although currently no such therapies are available. Previously, in utero administration of recombinant EDA was found to permanently correct the phenotype of newborn affected mice [58]. More recently, postnatal intravenous administration of soluble recombinant EDA to affected dogs resulted in normalization of adult teeth, improved ability to sweat, restoration of normal lacrimation, and decreased respiratory infection, due to improved mucociliary clearance from correction of glandular deficit in the trachea and bronchi [59,60].
Hypohidrotic Ectodermal Dysplasia with Immunodeficiency (EDA–ID; MIM 300291); Hypohidrotic Ectodermal Dysplasia with Immunodeficiency with Osteopetrosis and Lymphoedema (MIM 300301)
Both of these conditions are caused by mutations in the IKKγ gene, also known as NEMO. The observation of unusually severe recurrent infections in a small subset of patients with otherwise typical HED features led to the suggestion that there may be a specific syndrome of HED and immunodeficiency (EDA–ID). The EDA–ID syndrome was first reported in a boy with miliary tuberculosis [61]. The second reported patient had recurrent life-threatening infections caused by Pseudomonas aeruginosa, Mycobacterium avium and cytomegalovirus [62]. A third child had a milder phenotype with repeated infections due to Staphylococcus aureus and Streptococcus pneumoniae [63]. Three siblings from a different kindred had recurrent severe infections with Strep. pneumoniae, with impaired response to polysaccharide antigens [64]. All of these patients were male, suggesting X-linked inheritance. Further cases have extended our knowledge of the phenotype, and severe life-threatening or recurrent bacterial infections have been reported in the lower respiratory tract, skin, soft tissues, bones and gastrointestinal tract, as well as meningitis and septicaemia in early childhood.
Overall, the causative pathogens have most often been Gram-positive bacteria (Strep. pneumoniae and Staph. aureus) followed by Gram-negative bacteria (Pseudomonas spp. and Haemophilus influenzae) and mycobacteria. Most patients have severe hypogammaglobulinaemia with low serum IgG levels and varied levels of other immunoglobulin isotypes (IgA, IgM and IgE) [4]. Some patients have massively elevated IgM levels [20,22,65], and an impaired antibody response to polysaccharides is the most consistent feature of this condition [4]. Impaired natural killer (NK) cell activity is reported in some, but not all, patients with EDA–ID [66,67]; the degree and range of immunological abnormalities seen may relate to the type of NEMO mutation involved. Treatment of one such case with allogenic transplantation of umbilical cord blood resulted in the resolution of the eczematous eruption, although it is not clear if the defect in cellular immunity also resolved [68].
Autosomal Dominant Anhidrotic Ectodermal Dysplasia with T-Cell Immunodeficiency (MIM 612132)
The importance of these pathways was further emphasized when a mutation was identified in a 7-year-old boy with autosomal dominant anhidrotic ectodermal dysplasia and T-cell immunodeficiency. A 94G-T transversion resulting in a serine 32 to isoleucine (S32I) change in the IkBa gene (inhibitor of kappa light chain gene enhancer in B cells alpha), also known as NFKBIA (nuclear factor of kappa light chain gene enhancer in B cell inhibitor alpha) was found [69]. Ser32 is a key phospho-acceptor site of IκBα, and is conserved in the other two IκB proteins. The mutation appeared to be a de novo event. The patient was born to unrelated parents. From 2 months of age he had chronic diarrhoea, recurrent bronchopneumonitis, hepatosplenomegaly and failure to thrive. A diagnosis of ectodermal dysplasia with immunodeficiency was made at the age of 3 years on the basis of a dry rough skin, moderately sparse scalp hair and conical teeth. The patient had no other overt developmental defects. This patient was treated with a successful bone marrow transplant, although occasional immunoglobulin substitution was required post transplant [70]. Three other cases, including one female, have been described with similar immunodeficiency, presenting with recurrent infections, abnormal teeth, coarse, wrinkled or dry skin and, in some, thin hair and recessed hairline [71–73].
Familial Incontinentia Pigmenti (Incontinentia Pigmenti Type 2; MIM 308300)
Please see Chapter 130 for a full discussion of this condition.
References
1 Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998;16:225–60.
2 Kaufman CK, Fuchs E. It’s got you covered. NF-kappaB in the epidermis. J Cell Biol 2000;149:999–1004.
3 Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination. The control of NF-[kappa]B activity. Annu Rev Immunol 2000;18:621–63.
4 Smahi A, Courtois G, Rabia SH et al. The NF-kappaB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet 2002;11:2371–5.
5 Gerondakis S, Grossmann M, Nakamura Y et al. Genetic approaches in mice to understand Rel/NF-kappaB and IkappaB function: transgenics and knockouts. Oncogene 1999;18:6888–95.
6 Descargues P, Sil AK, Karin M. IKKalpha, a critical regulator of epidermal differentiation and a suppressor of skin cancer. EMBO J 2008;27(20):2639–47.
7 Ferguson BM, Brockdorff N, Formstone E et al. Cloning of Tabby, the murine homolog of the human EDA gene: evidence for a membrane-associated protein with a short collagenous domain. Hum Mol Genet 1997;6:1589–94.
8 Kere J, Srivastava AK, Montonen O et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 1996;13:409–16.
9 Drogemuller C, Distl O, Leeb T. Partial deletion of the bovine ED1 gene causes anhidrotic ectodermal dysplasia in cattle. Genome Res 2001;11:1699–705.
10 Drogemuller C, Peters M, Pohlenz J et al. A single point mutation within the ED1 gene disrupts correct splicing at two different splice sites and leads to anhidrotic ectodermal dysplasia in cattle. J Mol Med 2002;80:319–23.
11 Casal ML, Scheidt JL, Rhodes JL et al. Mutation identification in a canine model of X-linked ectodermal dysplasia. Mamm Genome 2005;16(7):524–31.
12 Yan M, Wang LC, Hymowitz SG et al. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science 2000;290:523–7.
13 Chen Y, Molloy SS, Thomas L et al. Mutations within a furin consensus sequence block proteolytic release of ectodysplasin-A and cause X-linked hypohidrotic ectodermal dysplasia. Proc Natl Acad Sci USA 2001;98:7218–23.
14 Han D, Gong Y, Wu H et al. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur J Med Genet 2008;51(6):536–46.
15 Barsh G. Of ancient tales and hairless tails. Nat Genet 1999;22:315–16.
16 Headon DJ, Overbeek PA. Involvement of a novel TNF receptor homologue in hair follicle induction. Nat Genet 1999;22:370–4.
17 Headon DJ, Emmal SA, Ferguson BM et al. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001;414:913–16.
18 Bal E, Baala L, Cluzeau C et al. Autosomal dominant anhidrotic ectodermal dysplasias at the EDARADD locus. Hum Mutat 2007;28(7):703–9.
19 Monreal AW, Ferguson BM, Headon DJ et al. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet 1999;22(4):366–9.
20 Doffinger R, Smahi A, Bessia C et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 2001;27:277–85.
21 Smahi A, Courtois G, Vabres P et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000;405:466–72.
22 Zonana J, Elder ME, Schneider LC et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 2000;67:1555–62.
23 Kojima T, Morikawa Y, Copeland NG et al. TROY, a newly identified member of the tumor necrosis factor receptor superfamily, exhibits a homology with Edar and is expressed in embryonic skin and hair follicles. J Biol Chem 2000;275:20742–7.
24 Eby MT, Jasmin A, Kumar A et al. TAJ, a novel member of the tumor necrosis factor receptor family, activates the c-Jun N-terminal kinase pathway and mediates caspase-independent cell death. J Biol Chem 2000;275:15336–42.
25 Naito A, Yoshida H, Nishioka E et al. TRAF6-deficient mice display hypohidrotic ectodermal dysplasia. Proc Natl Acad Sci USA 2002;99:8766–71.
26 Blake PW, Toro JR. Update of cylindromatosis gene (CYLD) mutations in Brooke–Spiegler syndrome: novel insights into the role of deubiquitination in cell signaling. Hum Mutat 2009;30(7):1025–36.
27 Trompouki E, Hatzivassiliou E, Tsichritzis T et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 2003;424(6950):793–6.
28 Kovalenko A, Chable-Bessia C, Cantarella G et al. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 2003;424(6950):801–5.
29 Brummelkamp TR, Nijman SM, Dirac AM et al. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 2003;424(6950):797–801.
30 Hutti JE, Shen RR, Abbott DW et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKK-epsilon promotes cell transformation. Molec Cell 2009;34:461–72.
31 Zenker M, Mayerle J, Lerch MM et al. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson–Blizzard syndrome). Nat Genet 2005;37(12):1345–50.
32 Thurnam J. Two cases in which the skin, hair and teeth were very imperfectly developed. Med Chir Trans 1848;31:71–82.
33 Thadani KI. A toothless type of man. J Hered 1921;12:87–8.
34 Clouston H. The major forms of hereditary ectodermal dysplasia (with an autopsy and biopsies on the anhydrotic type). Can Med Assoc J 1939;40:1–7.
35 Arnold ML, Rauskolb R, Anton-Lamprecht I et al. Prenatal diagnosis of anhidrotic ectodermal dysplasia. Prenat Diagn 1984;4(2):85–98.
36 Weech A. Hereditary ectodermal dysplasia (congenital ectodermal defect). A report of two cases. Am J Dis Child 1929;37:766–90.
37 Baer ST, Coulson IH, Elliman D. Anhidrotic ectodermal dysplasia: an ENT presentation in infancy. J Laryngol Otol 1988;102:458–9.
38 Micali G, Cook B, Blekys I et al. Structural hair abnormalities in ectodermal dysplasia. Pediatr Dermatol 1990;7(1):27–32.
39 Vierucci S, Baccetti T, Tollaro I. Dental and craniofacial findings in hypohidrotic ectodermal dysplasia during the primary dentition phase. J Clin Pediatr Dent 1994;18:291–7.
40 Clarke A, Phillips DI, Brown R et al. Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch Dis Child 1987;62:989–96.
41 Reed WB, Lopez DA, Landing B. Clinical spectrum of anhidrotic ectodermal dysplasia. Arch Dermatol 1970;102:134–43.
42 Crawford PJ, Aldred MJ, Clarke A. Clinical and radiographic dental findings in X linked hypohidrotic ectodermal dysplasia. J Med Genet 1991;28:181–5.
43 Clauss F, Manière MC, Obry F et al. Dento-craniofacial phenotypes and underlying molecular mechanisms in hypohidrotic ectodermal dysplasia (HED): a review. J Dent Res 2008;87(12):1089–99.
44 Levin LS. Dental and oral abnormalities in selected ectodermal dysplasia syndromes. Birth Defects Orig Artic Series 1988;24:205–27.
45 Happle R, Frosch PJ. Manifestation of the lines of Blaschko in women heterozygous for X-linked hypohidrotic ectodermal dysplasia. Clin Genet 1985;27:468–71.
46 Executive and Scientific Advisory Boards of the National Foundation for Ectodermal Dysplasias, Mascoutah, Illinois. Scaling skin in the neonate: a clue to the early diagnosis of X-linked hypohidrotic ectodermal dysplasia (Christ–Siemens–Touraine syndrome). J Pediatr 1989;114(4 Part 1):600–2.
47 Clarke A. Hypohidrotic ectodermal dysplasia. J Med Genet 1987;24:659–63.
48 Butterworth T, Ladda R. Clinical Genodermatology. Westpoint, CT: Praeger, 1981, pp. 208–17.
49 Coston GN, Salinas CF. Speech characteristics in patients with hypohidrotic ectodermal dysplasia. Birth Defects Orig Artic Series 1988;24:229–34.
50 Wright JT, Finley WH. X-linked recessive hypohidrotic ectodermal dysplasia. Manifestations and management. Ala J Med Sci 1986;23(1):84–7.
51 Soderholm AL, Kaitila I. Expression of X-linked hypohidrotic ectodermal dysplasia in six males and in their mothers. Clin Genet 1985;28:136–44.
52 Freire-Maia N, Pinheiro M. Ectodermal Dysplasias: a Clinical and Genetic Study. New York: Alan R. Liss, 1984.
53 Sybert VP. Early diagnosis in the ectodermal dysplasias. Birth Defects Orig Artic Series 1988;24:277–8.
54 Nowak AJ. Dental treatment for patients with ectodermal dysplasias. Birth Defects Orig Artic Series 1988;24:243–52.
55 Guckes AD, Brahim JS, McCarthy GR et al. Using endosseous dental implants for patients with ectodermal dysplasia. J Am Dent Assoc 1991;122(11):59–62.
56 Myer CM 3rd. The role of an otolaryngologist in the care of ectodermal dysplasia. Pediatr Dermatol 1987;4:34–5.
57 Myer CM 3rd. Otolaryngologic manifestations of the ectodermal dysplasias – clinical note. Int J Pediatr Otorhinolaryngol 1986;11:307–10.
58 Gaide O, Schneider P. Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat Med 2003;9:614–18.
59 Casal ML, Lewis JR, Mauldin EA et al. Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am J Hum Genet 2007;81(5):1050–6.
60 Mauldin EA, Gaide O, Schneider P et al Neonatal treatment with recombinant ectodysplasin prevents respiratory disease in dogs with X-linked ectodermal dysplasia. Am J Med Genet A 2009;149A(9):2045–9.
61 Frix CD 3rd, Bronson DM. Acute miliary tuberculosis in a child with anhidrotic ectodermal dysplasia. Pediatr Dermatol 1986;3:464–7.
62 Sitton JE, Reimund EL. Extramedullary hematopoiesis of the cranial dura and anhidrotic ectodermal dysplasia. Neuropediatrics 1992;23:108–10.
63 Abinun M, Spickett G, Appleton AL et al. Anhidrotic ectodermal dysplasia associated with specific antibody deficiency. Eur J Pediatr 1996;155:146–7.
64 Schweizer P, Kalhoff H, Horneff G et al. Polysaccharide specific humoral immunodeficiency in ectodermal dysplasia. Case report of a boy with two affected brothers. Klin Padiatr 1999;211:459–61.
65 Jain A, Ma CA, Liu S et al. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2001;2:223–8.
66 Orange JS, Brodeur SR, Jain A et al. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J Clin Invest 2002;109:1501–9.
67 Dupuis-Girod S, Corradini N, Hadj-Rabia S et al. Osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency in a boy and incontinentia pigmenti in his mother. Pediatrics 2002;109(6):e97.
68 Minakawa S, Takeda H, Nakano H et al. Successful umbilical cord blood transplantation for intractable eczematous eruption in hypohidrotic ectodermal dysplasia with immunodeficiency. Clin Exp Dermatol 2009;34(7):e441–2.
69 Courtois G, Smahi A, Reichenbach J et al. A hypermorphic IκBα mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and t-cell immunodeficiency. J Clin Invest 2003;112:1108–15.
70 Dupuis-Girod S, Cancrini C, Le Deist F et al. Successful allogeneic hemopoietic stem cell transplantation in a child who had anhidrotic ectodermal dysplasia with immunodeficiency. Pediatrics 2006;118(1):205–11.
71 Janssen R, van Wengen A, Hoeve MA et al. The same I-kappa-B-alpha mutation in two related individuals leads to completely different clinical symptoms. J Exp Med 2004;200:559–68.
72 Lopez-Granados E, Keenan JE, Kinney MC et al. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat 2008;29:861–8.
73 McDonald DR, Mooster JL, Reddy M et al. Heterozygous N-terminal deletion of I-kappa-B-alpha results in functional nuclear factor kappa-B haploinsufficiency, ectodermal dysplasia, and immune deficiency. J Allergy Clin Immunol 2007;120:900–7.
Transcription Factors, Homeobox Genes: Major Regulators of Gene Expression
TP63-Related Phenotypes: Overview of Molecular Pathway
The p53 gene family is a key regulator of the cell cycle and is mutated in more than 50% of human cancers. p63 and p73 are recently discovered [1–5], related genes that share high amino acid identity with p53. The role of p63 and p73 in human cancers has been extensively studied, but neither molecule is believed to play a significant role in tumorigenesis. Both p63 and p73 are distinct from p53 in that they each have a C-terminal protein–protein interacting motif (sterile α motif – SAM domain) that is not present in p53. The p63 and p73 genes also differ from p53 in that they can each encode several different isoforms by utilizing two different transcription initiation sites (for review see ref. 6). The expression of p63 is more restricted than the ubiquitous nature of p53 expression and is restricted to the embryonic ectoderm and the basal regenerative layers of epithelial tissues in adults (skin, cervix, tongue, oesophagus, mammary glands, prostate and urothelium [5]). p63 −/− mice die at birth and have truncation of limbs (especially the forelimbs, with complete absence of phalanges and carpals) and absence of ectodermal derivatives including the epidermis and appendages (whiskers, hair, etc.), the prostate, lacrimal, breast and urothelial tissues [7,8]. Limb defects are best explained by a failure of the apical ectodermal ridge to develop [8].
Mutations in the human p63 gene, TP63, have now been identified in six distinct human phenotypes, all of which have ectodermal dysplasia as a key feature. Some genotype–phenotype correlation is possible in that there is clustering of mutations in some of the phenotypes to specific sites of the p63 molecule (for review see ref. 6). TP63 mutations account for most cases of ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC syndrome). In an authoritative paper, van Bokhoven et al. [9] were able to demonstrate mutations in 40 out of 43 families with EEC; all but one of these mutations were sited within the DNA-binding domain, and five amino acid residues accounted for 75% of all mutations [9]. In ankyloblepharon-ectodermal dysplasia-clefting syndrome (AEC syndrome, also known as Hay–Wells syndrome), in which the limb abnormalities are absent or minimal, mutations have been exclusively detected within the SAM domain and are associated with complex gain-of-function as well as loss- and change-of-function effects [9]. In limb-mammary syndrome (LMS), TP63 frameshift mutations leading to truncations of p63 protein have been reported in exon 13 in two unrelated patients [9] and an N-terminal mutation was found in a further family [6]. Mutations in acro-dermato-ungual–tooth syndrome (ADULT syndrome) have yielded interesting insights in that the first mutation was identified in exon 3′, which is only expressed in the transactivating (TA) isotypes of p63 and causes an amino acid substitution outside the DNA-binding domain [10]. A subsequent report has demonstrated a mutation that confers significant transactivation activity on ΔN-p63γ, an otherwise inert isoform of p63 [6]. Non-syndromic split hand-split foot syndrome (SHFM) is a genetically heterogeneous group of conditions, but some cases (possibly around 10%) are attributable to p63 mutations [9]. Some of these mutations seem specific for SHFM, but others underlie both EEC and SHFM [6]. It is therefore understandable that of the six known types of SHFM, ectodermal dysplasia features are most commonly seen in SHFM4, which shares the same gene as EEC, while SHFM3, which maps to 10q24, has only dental and nail findings [11].
The TP63 gene product can act as an activator or a repressor, and mutations in the 5′ end of TP63 in AEC and Rapp–Hodgkin syndrome show loss of activator function, and even dominant-negative activity [12,13]. While clustering of mutations is determined in part by the characteristics of the mutated residue, whether it is a CpG site, etc., the degree of clustering in this group of conditions suggests that each condition has a specific pathogenetic mechanism. This site specificity also presumably suggests that the p63 protein has several functions, each with a specific site, and that each of these functions can be disturbed in isolation from the others. However, overlapping phenotypes are not uncommon and further elucidation of TP63 may shed light on the role of this protein and its downstream pathway [14,15]. One target of p63 is PERP (p53 effector related to PMP22), a gene that has an effect on cell–cell adhesion and is a potential tumour suppressor gene [16,17]. PERP levels may vary in patients with the same TP63 mutation; therefore modifier genes and other proteins are likely to be involved in the phenotypic expression of TP63 mutations [18]. IKKα, discussed above, is a direct transcriptional target of ΔNp63α [19,20]. A mouse model may shed further light on the role of p63 in ectodermal dysplasia [21].
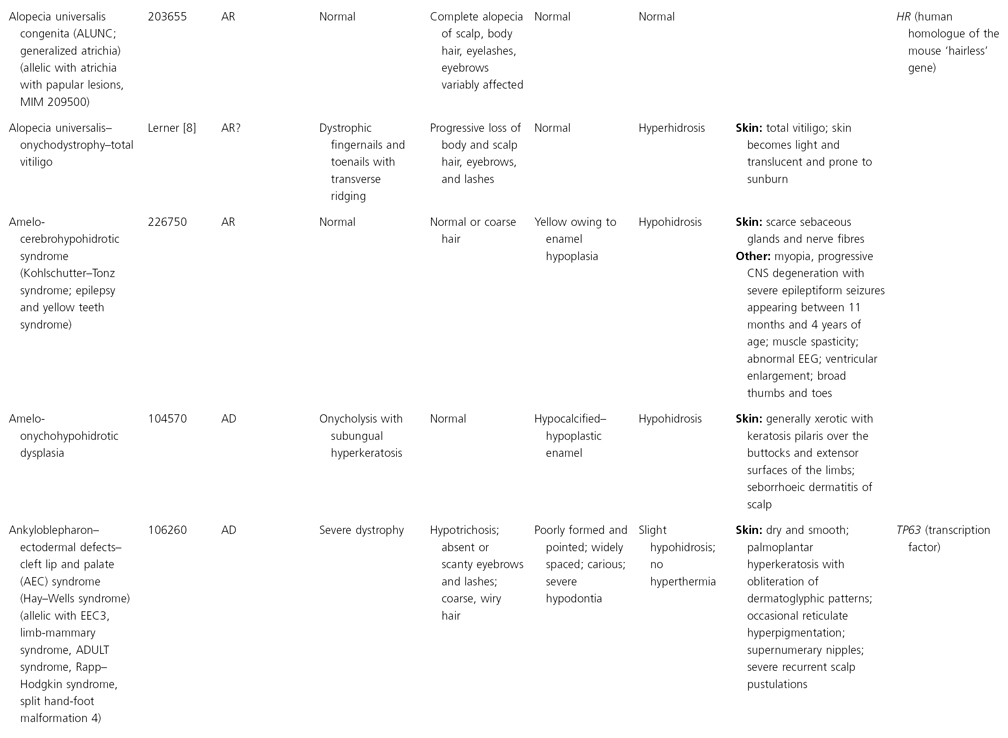
Ankyloblepharon-Ectodermal Dysplasia-Clefting Syndrome (AEC; Hay–Wells Syndrome; MIM 106260)
Definition.
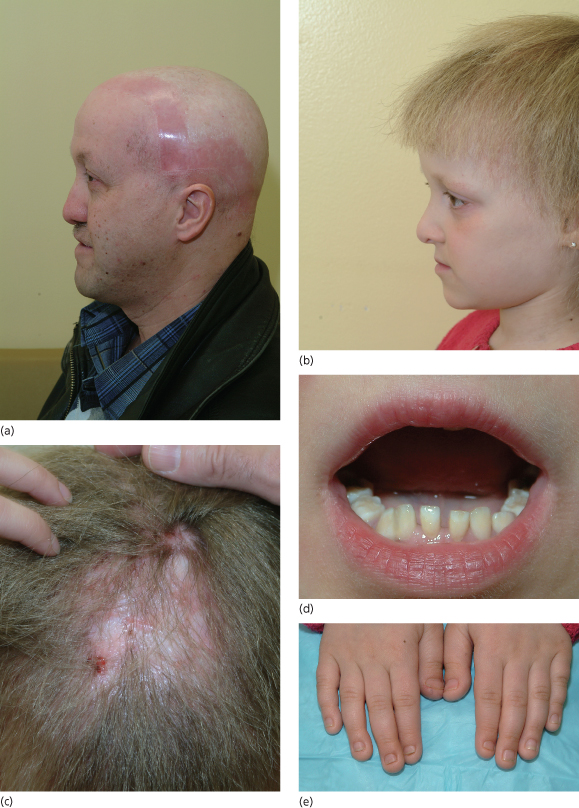
Ankyloblepharon-ectodermal dysplasia-clefting syndrome is characterized by cleft lip/palate, severe scalp erosions and abnormalities of the epidermal appendages including hypotrichosis, hypodontia, absent or dystrophic nails and mild hypohidrosis. One distinctive feature is ankyloblepharon filiforme adnatum, partial-thickness fusion of the eyelid margins. Recently, mutations in TP63 have also been identified in Rapp–Hodgkin syndrome (MIM 129400) (Fig. 127.8) [22], demonstrating that this disorder is allelic with AEC syndrome. Clinical distinction between these two disorders is probably not warranted.
Fig. 127.8 (a,b) A father and daughter with Rapp–Hodgkin syndrome (AEC). Both have history of cleft palate. Note the typical facies (high forehead, hypoplastic maxilla and thin upper lip) and brittle, wiry hair in childhood, which can progress to alopecia in adulthood. The father was previously wearing a hairpiece. (c) Scalp dermatitis with alopecia seen in Rapp–Hodgkin syndrome (AEC). (d,e) Hypodontia and nail dysplasia in Rapp–Hodgkin syndrome (AEC).
Courtesy of Dr Jean Bernard.
History.
In 1976, Hay and Wells described seven patients from four families with an inherited disorder characterized by congenital filiform fusion of the eyelids, dysplasia of the epidermal appendages and cleft lip/palate. Five of these original seven patients had ankyloblepharon filiforme adnatum and one had small nodules removed from her eyelids as a child, presumably remnants of spontaneously lysed ankyloblepharon [23].
Pathology.
Hair shafts are thin and atrophic, and show various defects, including fractures of the cuticle, bent shafts, trichoclasis, trichorrhexis nodosa, pili canaliculi, pili annulati, pili triangulati and pili torti, none of which is specific for the disorder [24,25]. Dyspigmentation of the hair is relatively common, with pigment variation both between and within hairs; pigment may be normal, clumped or nearly absent [25].
Skin biopsy of involved scalp tissue shows a thin granular layer and stratum corneum [26]. Biopsies from clinically ‘normal’ skin show mild hyperkeratosis and papillomatosis, epidermal atrophy and pigment incontinence, as well as a prominent superficial perivascular plexus with minimal to mild perivascular lymphocyte infiltrates [25]. Hair follicles are reduced in size and arrector pili muscles appear hypertrophic [26]. Sweat stimulation tests reveal a patchy loss of sweat glands over most of the body [23].
Clinical Features
Hair
Scalp hair is wiry, coarse and sparse; alopecia is common. The eyebrows and eyelashes are almost always short, brittle and sparse or absent. Body, pubic and axillary hair may be sparse or absent [23,24,27]. Some may also display scarring alopecia or ‘spun-glass’ uncombable hair [28].
Teeth
Hypodontia is common. Those teeth that are present are frequently small, conical and discoloured with white or yellow spots secondary to enamel hypoplasia [23,24,27,29,30]. Maxillary and mandibular first molars are the most likely of the permanent teeth to be present, while maxillary incisors and canines tend to be absent, probably due to the alveolar cleft defect [30].
Nails
Nail abnormalities are variable even within an individual and include distal hypoplasia and thickened, hyperconvex plates. Complete absence of nails is a frequent finding [23]. Chronic paronychia has been reported [24].
Sweat Glands
Decreased sweat production and heat intolerance is described by a significant number of individuals but hyperpyrexia is not a problem [27,28]. Sweat pores are reduced in a number in affected individuals [23].
Skin
At birth, over three-quarters of affected newborns have red, eroded, peeling skin like a collodion membrane (Fig. 127.9) [27] or may present with erythroderma [31]. Erosions are most prominent over the scalp [28]. These symptoms resolve over the first few weeks and the underlying skin is dry (Fig. 127.10). Erosions may heal with residual scarring, in a cribriform, reticulate, stellate or punctate pattern [28]. Over two-thirds of individuals have chronic problems with severe recurrent scalp erosions and scalp infections, which are a major feature of AEC syndrome (Fig. 127.11) [27]. Palmoplantar keratoderma was reported in four of the original seven patients described by Hays and Wells [23]. It is not a common finding in affected children but may be more pronounced in adults. Both hypopigmentation and hyperpigmentation can occur, commonly in a reticulate pattern in intertriginous areas; this may progress with age [28].
Fig. 127.9 Peeling, red, parchment-like skin in a newborn with AEC syndrome.
Courtesy of Dr Virginia Sybert.
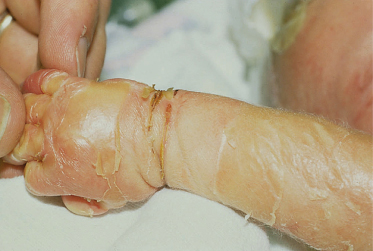
Fig. 127.10 The parchment skin resolves over the first few weeks of life and the underlying skin is dry as seen in this 1-month-old baby with AEC syndrome.
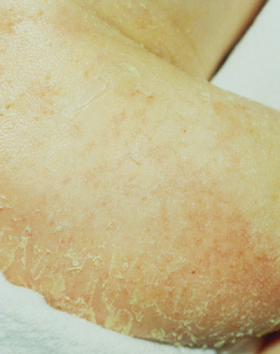
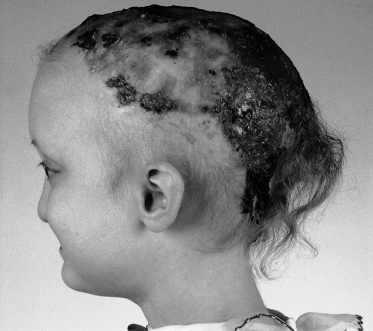
Fig. 127.11 Severe scalp erosions and extensive granulation tissue in a 5-year-old girl with AEC syndrome.
Courtesy of Dr Virginia Sybert and Dr Mark Stephan.
Other Ectodermal Structures
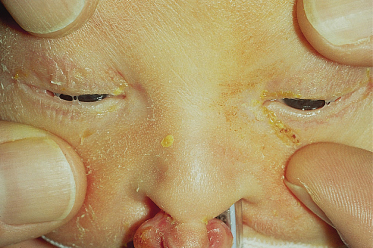
Ankyloblepharon filiforme adnatum (strands of epithelial tissue between the eyelids) are a cardinal feature of the disorder but are noted in only 70% of patients (Fig. 127.12) [23,24,27]. The strands may lyse spontaneously and may be difficult to detect. Lacrimal duct atresia or obstruction occurs in over half of affected individuals [27]. Supernumerary nipples may be present [24,27]. Nutritional issues are common, which are not specifically related to cleft palate and lip. One-quarter of patients require gastrostomy placement, and nutritional supplements at some point in time are required in two-thirds of patients. Low birthweight is common, although birth length is unaffected. Weight issues resolve with time, but AEC patients have significantly lower height-for-age than the reference population [32].
Fig. 127.12 Ankyloblepharon filiforme adnatum (strands of epithelial tissue between the eyelids) in a newborn with AEC syndrome.
Courtesy of Dr Virginia Sybert.
Craniofacial Features
Typical craniofacial features include a broadened nasal bridge and maxillary hypoplasia [33]. The ears may be small and low-set with deformities of the auricle [27]. The ear canals may be webbed and abnormally shaped [24]. Cleft lip is a variable feature but cleft palate is seen in most individuals [23–27].
Other Abnormalities
Other features seen occasionally in AEC syndrome include cutaneous syndactyly of the second and third toes, hypospadias and vaginal dryness and erosions [27].
Prognosis.
Abnormalities of the external ear canals and palate frequently cause problems with chronic otitis media and secondary hearing loss. Atresia of the lacrimal duct can lead to excessive lacrimation, chronic conjunctivitis and photophobia. Scalp erosions and chronic scalp infections may be severe enough to warrant surgical intervention with skin engraftment [27]. Prenatal diagnosis of cleft palate/lip by ultrasound may be possible in affected families [34].
Differential Diagnosis.
Curly hair-ankyloblepharon-nail dysplasia syndrome (CHANDS) is a rare autosomal recessive ectodermal dysplasia with curly, kinky hair, hypoplastic nails and the defining feature of ankyloblepharon. It can be distinguished from AEC by absence of cleft palate and lack of typical craniofacial features [35,36]. In the newborn period, the eroded, peeling skin seen in AEC syndrome may be mistaken for epidermolysis bullosa [37].
Treatment.
Emollients are appropriate for the collodion-like membrane in the newborn. Neonates with AEC often have extremely fragile skin and they should be handled with extreme care. Neonatal intensive care nursing protocols such as those used for neonates with epidermolysis bullosa should be used. The ankyloblepharon filiforme adnatum may require surgical correction or may lyse spontaneously. The lacrimal duct atresia may be surgically correctable [38]. The scalp requires aggressive wound care and treatment with topical or systemic antibiotics as warranted [27]. Other abnormalities, such as cleft lip/palate, hypospadias and the maxillary hypoplasia, may be surgically corrected [33]. Teeth preservation and restoration are imperative [29].
Ectrodactyly-Ectodermal Dysplasia-Clefting Syndrome (EEC; MIM 12990)
Definition.
The main features of the EEC syndrome are ectrodactyly (spilt hand or foot deformity), cleft lip/palate, tear duct anomalies and abnormalities of the epidermal appendages including hypotrichosis, hypodontia, dystrophic nails and occasional hypohidrosis.
History.
The association of ectrodactyly, cleft lip/palate and ectodermal dysplasia was initially described by Rüdiger et al. [39], who recognized that this combination of defects represents a specific syndrome, termed EEC syndrome. Over 150 cases have subsequently been described [26].
Pathology.
Radiographs of hand or foot deformities show missing or hypoplastic metacarpals and metatarsals [40]. Scanning electron microscopic studies of hair shafts of affected individuals show longitudinal grooves, distorted bulbs and cuticular defects [41,42]. These findings can be seen in a number of other ectodermal dysplasias and are not specific to EEC syndrome.
Clinical Features
Hair
The scalp hair is fine and sparse, light-coloured and may be wiry in texture. Eyebrows and eyelashes are short, thin and sparse. Axillary, pubic and body hair may also be affected [39–41,43].
Teeth
Teeth may be small, abnormally shaped or missing [39–41,43]. Premature loss of secondary teeth is common, presumably owing to multiple caries from enamel hypoplasia.
Nails
The nail plates may be dystrophic, hypoplastic or completely absent even when there are no bony defects of the involved digit [43,44].
Sweat Glands
Sweating is usually normal but heat intolerance is noted by a few individuals [40,43,45].
Skin
Dry skin and hyperkeratosis, particularly of the lower extremities, are reported in some individuals [46,47]. Scalp dermatitis is seen rarely [41,47].
Other Ectodermal Structures
Atresia or hypoplasia of the lacrimal duct is seen in over 90% of affected individuals [26,46,48]. Secretions from the lacrimal gland may be diminished [48]. Nipple anomalies are reported in a few individuals [46].
Craniofacial Features
The nose may be broad, the chin pointed, and there may be minor variable ear anomalies, but the facies are not distinct. Cleft palate with or without cleft lip occurs in three-quarters of affected individuals and is a major feature of this disorder [46]. Choanal atresia has been reported [49].
Other Abnormalities
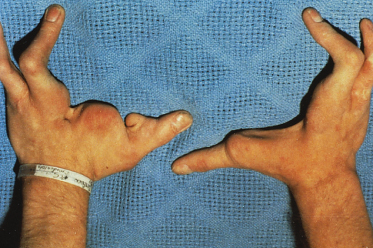
Ectrodactyly (lobster claw deformity) is a major feature of this disorder and occurs in over 90% of affected individuals (Fig. 127.13). About three-quarters of individuals with ectrodactyly have both hand and foot involvement [46]. Structural abnormalities of the genitourinary tract occur in about one-third of individuals, including cryptorchidism, hypospadias, hydronephrosis and hydroureters, renal agenesis, and duplication of the kidneys and collecting system [46]; the most common structural finding is megaureter [50,51]. Urinary abnormalities may be more common in those cases of EEC with an Arg227Gln TP63 mutation [52]. Abnormalities of the external genitalia have also been described [51]. Mental retardation is a variable and uncommon feature of the disorder, occurring in less than 10% of affected individuals [46], and may be limited to those with chromosomal deletions as part of a contiguous gene syndrome. Hearing loss occurs in about 15% of individuals [46]. It is uncertain whether this is primary or secondary to recurrent otitis media. Isolated growth hormone deficiency has been reported in one individual [53]. Endocrine anomalies such as hypogonadotropic hypogonadism, thyroid-stimulating hormone and prolactin deficiency have also been reported [54]. Cases of concomitant Hodgkin, non-Hodgkin and B-cell lymphoma in EEC have also been reported [54–56].
Fig. 127.13 Ectrodactyly of the hands in a young man with EEC syndrome.
Courtesy of Dr Virginia Sybert.
Prognosis.
A significant number of affected individuals experience excessive tearing, conjunctivitis and blepharitis as a result of lacrimal duct hypoplasia. Photophobia and corneal ulcers as well as corneal scarring and perforation may occur as a result of lacrimal gland hypoplasia [48]. Recurrent urinary tract infections, both symptomatic and asymptomatic, may be a problem in individuals with genitourinary anomalies [50].
Differential Diagnosis.
A few other ectodermal dysplasias involve limb abnormalities and cleft palate/lip. Although clefting is not a constant feature, odontotrichomelic syndrome may be differentiated by severe tetramelic reductions and autosomal recessive mode of inheritance [26]. Other rare syndromes, such as Martinez syndrome, Zlotogora–Ogur syndrome and Rosselli–Gulienetti syndrome, can be differentiated from EEC by specific limb abnormalities and mode of inheritance (see Table 127.1).
Treatment.
Treatment involves surgical correction of the cleft lip/palate, lacrimal duct and limb defects and genitourinary abnormalities as indicated. DNA-based prenatal diagnosis is available for selected families in which the gene defect is known.
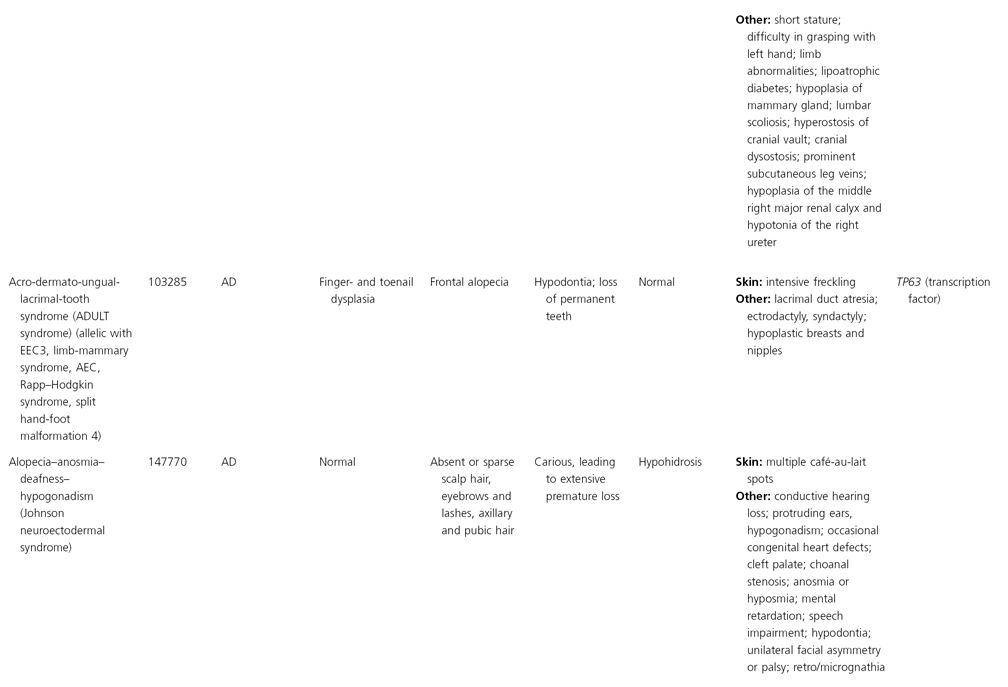
Acro-Dermato-Ungual-Tooth (ADULT; MIM 103285)
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


