AD, autosomal dominant; AR, autosomal recessive; XD, X-linked dominant.
On a pathogenetic basis, the bulk of erythrokeratodermas are due to genetic modifications of the gap junction protein family, the connexins. Gap junctions are clusters of intercellular plasma membrane channels connecting the cytoplasm of neighbouring cells and allowing free transfer of small molecules of less than 1 kDa. Gap junctions are essential for intercellular communication and thus for control of cell fate and tissue homeostasis. Connexins are components of connexons, which form gap junctions. These intercellular channels are formed by the docking of two connexons consisting of six connexins. All the connexins within a connexon can be the same (homomeric) or different (heteromeric); and the two connexons docking together can be identical (homotypic junctions) or different (heterotypic junctions). In the epidermis, nine different connexins (Cx) are expressed. In the basal cell layer, Cx26 is the predominant connexin, while in the suprabasal compartment the most expressed connexins are Cx43, Cx31, Cx30.3 and Cx37 (see Table 122.2).
Table 122.2 Synopsis of genodermatoses with erythrokeratotic or erythrosquamous component
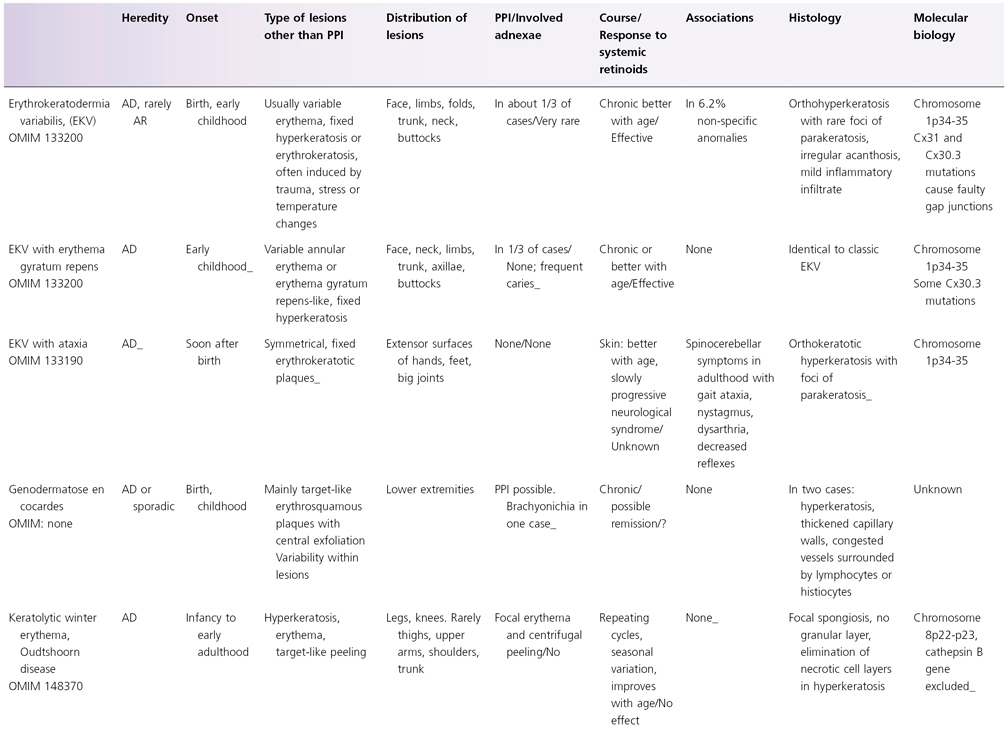
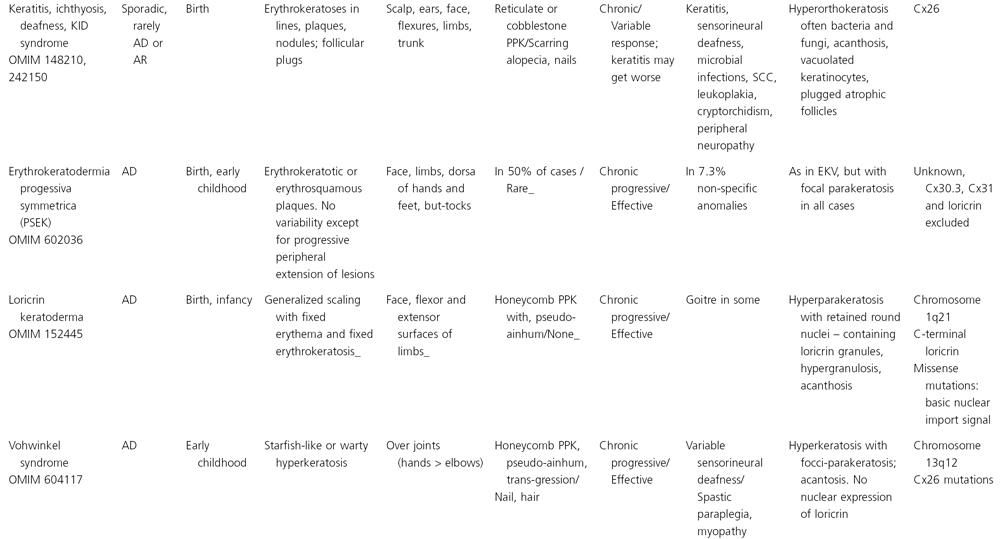
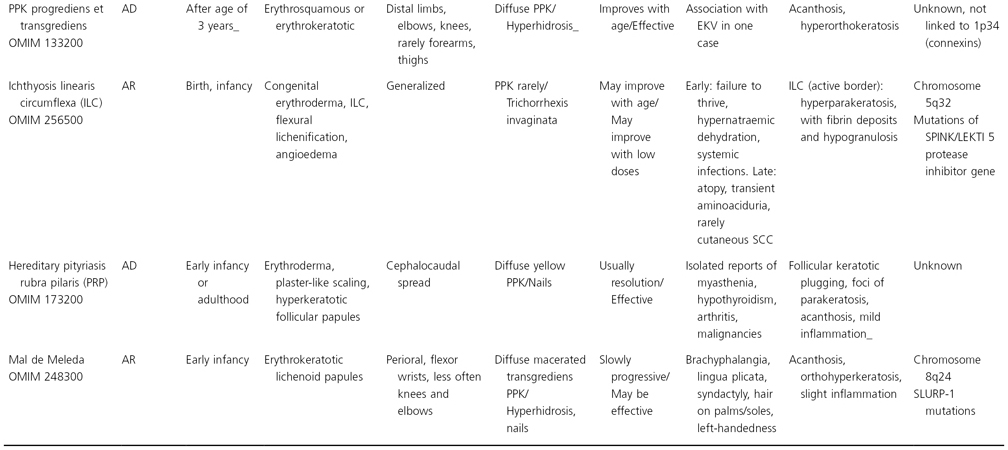
PPI, palmoplantar involvement; AD, autosomal dominant; AR, autosomal reccesive; Cx, connexin; PPK, palmoplantar keratoderma; SCC, squamous cell carcinoma.
*In two members of an Iranian family EKV and neurological anomalies appeared in early life and rapidly evolved toward death.
Keratitis-Ichthyosis-Deafness Syndrome
Syn.
Atypical ichthyosiform erythrokeratoderma
History.
The keratitis-ichthyosis-deafness (KID) syndrome was initially delineated by Burns [4]. Schnyder rediscovered KID syndrome in the 1960s [2,3] as a new neurocutaneous syndrome characterized by a partial PSEK, neurosensory deafness, myopathy and peripheral neuropathy. Similar cases were described by others [5–7], and Skinner et al. coined the term KID syndrome [8]. Ichthyosis hystrix of Rheydt also is probably a case of KID syndrome as the patient suffered from deafness. Additionally, the so-called HID (hystrix-like ichthyosis with deafness) syndrome and the Bart–Pumphrey syndrome were confirmed to share the same genetic mutations and represent clinical variants or variable expression of KID syndrome.
Epidemiology.
Over 100 cases have been reported in the literature, mostly sporadic, rarely of autosomal dominant or recessive inheritance.
Pathogenesis.
The KID syndrome is due to connexin 26 mutations (GJB2) [9]. A case bearing a connexin 30 mutation (GJB6) has been claimed to correspond to KID syndrome with atrichia [10]. In our view, such cases should be diagnosed as Clouston syndrome.
The dermatopathological changes are non-specific with acanthosis and hyperkeratosis, similar to the papillated epidermal hyperplasia seen in PSEK and EKV.
Clinical Features.
The phenotype of KID syndrome is variable, which can be understood in the context of the wide clinical spectrum of allelic connexin 26 defects, which includes Vohwinkel syndrome, hystrix-like-ichthyosis-deafness (HID) syndrome [11], the Bart–Pumphrey syndrome, and autosomal recessive type1 and autosomal dominant type 3 deafness. The HID phenoytpe differs from KID by its marked generalized spiky and cobblestone-like hyperkeratoses and only a mild palmoplantar keratoderma. The Bart–Pumphrey syndrome is characterized by sensorineural deafness, knuckle pads, palmoplantar keratoderma and leukonychia.
Most KID syndrome patients have skin manifestations at birth, and all develop skin changes by 1 year of age [12]. Neonates present with erythroderma and/or a vernix-like hyperkeratosis. The neonatal erythroderma rapidly clears and symmetric erythematous and verrucous plaques develop in the first months or years of life, and progress until puberty. The fixed irregularly shaped, erythematous keratotic plaques typically involve the face and are accentuated over joints, somewhat reminiscent of the starfish-shaped keratoses of Vohwinkel syndrome. The lesions have a particular grainy or cobblestone surface (Fig. 122.1), which is also found on the palmoplantar keratoderma.
Fig 122.1 Keratitis-Ichthyosis-Deafness (KID) syndrome with its characteristic verrucous widespread hyperkeratosis and deafness. Note that the clinical aspect is keratodermic, not ichthyotic.
Upper panel courtesy of Dr M. Harms, Geneva.
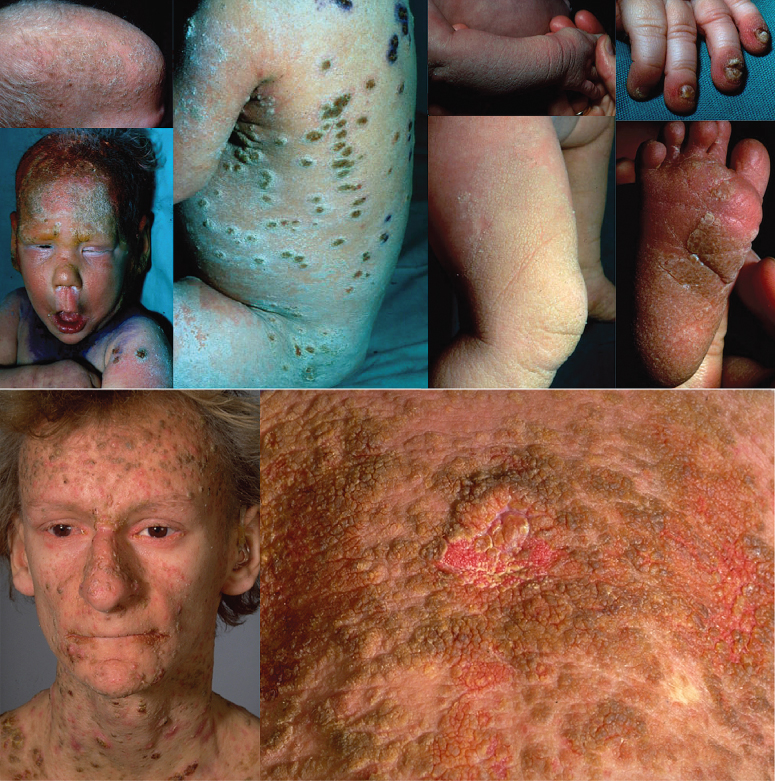
Most patients with KID syndrome have absent or sparse scalp hair, and may also have abnormal eyelashes, eyebrows, axillary and pubic hair. Alopecia may be present at birth, or may progress over time secondary to recurrent infections and subsequent scarring. Other features are follicular keratoses with spiny projections, verrucous hyperkeratoses, perioral furrowing, leuconychia and nail dystrophy. Occasionally, nails may be absent at birth [13]. Teeth may be small or malformed [14] with a tendency for increased dental caries. There are rare reports of oral leucokeratosis and scrotal tongue [1]. Lazic et al. have reported a variant with a specific connexin 26 mutation in which fissured lips and gingival erythema are prominent clinical features [15].
Bilateral non-progressive, congenital sensorineural hearing defects are present that range in severity from mild to profound [16]. In contrast with the congenital hearing impairment, ocular changes may not be observed until the second or third decade of life and are usually progressive. Photophobia may uncommonly be present at birth [12]. Early ocular findings include photophobia, tearing and conjunctivitis. Over time, patients develop a typical, progressive proliferating vascular keratitis [17], which leads to decreased visual acuity and potential blindness.
For reasons that remain poorly understood, approximately one-half of reported patients have problems with severe and recurrent cutaneous bacterial and fungal infections [12,18]. Finally, close to 20% of patients develop cutaneous or oral squamous cell carcinomas [19].
Prognosis.
There is a chronic disease course. Recurrent squamous cell carcinomas may reduce life expectancy.
Differential Diagnosis.
Hidrotic ectodermal dysplasia (Clouston syndrome), also due to connexin 30 mutations, shows more severe ectodermal defects, notably hair and nail defects. Likewise, atrichia with KID-like symptoms due to a dominant connexin 30 mutation has been described [10]. The allelic Vohwinkel syndrome, HID syndrome [11], the Bart–Pumphrey syndrome or autosomal recessive type 1 and autosomal dominant type 3 deafness can be distinguished based on clinical features.
Treatment.
The treatment is challenging, and emollients and keratolytic agents are of limited value. Oral retinoids may improve hyperkeratoses but lead to eye irritation.
Erythrokeratodermia Variabilis Mendes Da Costa
Syn.
Keratosis rubra figurata, erythrokeratodermia figurata variabilis, Mendes da Costa disease, erythrokeratodermia figurata variabilis type 1
History.
DeBuy Wenniger [20] reported the disease first in 1907, followed in 1922 by Rille [21] (keratosis rubra figurata) and Jeanselme [22]. In 1925, Mendes da Costa [23] coined the term ‘Erythro- et Keratodermia variabilis in a mother and daughter’ (EKV) to describe a heritable disorder characterized by fixed hyperkeratotic plaques and by erythematous areas with outlines ‘like the boundary lines of seacoasts’ that moved ‘in the course of an hour’.
Epidemiology.
Erythrokeratodermia variabilis Mendes da Costa accounts for about two-thirds of all EKV. This rare genodermatosis has been reported in all races with an equal gender incidence. The pattern of inheritance, studied in some large families, was reported to be autosomal dominant [24,25]. However, some very rare cases of autosomal recessive inheritance have also been described [26]. Its estimated incidence in Switzerland is about 1:500,000.
Pathogenesis.
Cases of EKV are due to germline mutations in the gap junction protein beta 3 and beta 4 genes (GJB3, GJB4) that code for the gap junction proteins connexin 31 (Cx31) and 30.3 (Cx30.3), respectively [27,28]. About half of all EKV are due to mutations in Cx31, one-third to mutations in Cx30.3 [29,30]. Connexin 30.3 mutations cause less severe phenotypes generally and account also for EKV Cram–Mevorah (see below). On a molecular level, Cx31 has been suggested to interact with Cx43 (oculodentodigital dysplasia) and 30.3 [31]. In most cases of EKV, mutations are thought to cause disease by a dominant-negative mechanism disturbing intercellular coupling of connexons and trafficking. More recently, a family has been described with EKV showing autosomal recessive inheritance being caused by a homozygous mutation in the GJB3 gene [32].
Pathology.
The dermatopathological changes are non-specific. They show a compact hyperkeratosis with papillomatosis and acanthosis as well as a sparse, mononuclear perivascular infiltrate in the upper dermis [33–35]. Electron microscopy and immunohistological studies have shown inconsistent results, with a decrease in lamellar bodies or dense perinuclear tonofilament and keratohyalin granules [33–36].
Clinical Features.
Erythrokeratodermia variabilis Mendes da Costa has two morphologically different types of skin lesions: migratory erythematous areas and fixed hyperkeratotic plaques. Skin lesions are usually present at birth or develop during the first year of life but may rarely develop during late childhood or early adulthood [24,37–39]. The disease presents first as erythema and only with time do hyperkeratotic lesions appear. Although characteristically these components do not occur on the same skin area, hyperkeratotic plaques are often surrounded by erythema. The erythematous patches have irregular borders, occur at any site and migrate slowly over the body (Fig. 122.2). They last from hours to days, may cause slight pruritus or burning sensation, and may show some fine scaling. They are influenced by emotions, physical stressors such as temperature changes, friction or pressure, and hormonal changes. The fixed geographical, hyperkeratotic plaques develop primarily on the extremities, buttocks and face. They are well demarcated, range from red to yellowish-brown in colour and extend over the years. Erythrokeratodermia variabilis may flare under the influence of oral contraceptive pills or pregnancy and may improve after menopause [34]. Thus, the clinical features vary not only within a patient but also within the affected family members. Thus, there is considerable variability both within the patient over time and also between affected family members. Palmoplantar keratoderma, usually in the form of a fine erythematous scaling, may be associated [38–40], although severely affected patients may exhibit diffuse keratoderma (M. Williams and D. Hohl, unpublished observations).
Fig 122.2 Erythrokeratoderma variabilis Mendes da Costa due to mutated Cx 31 in a family followed over four generations. Lesions become more fixed and hyperkeratotic with age.
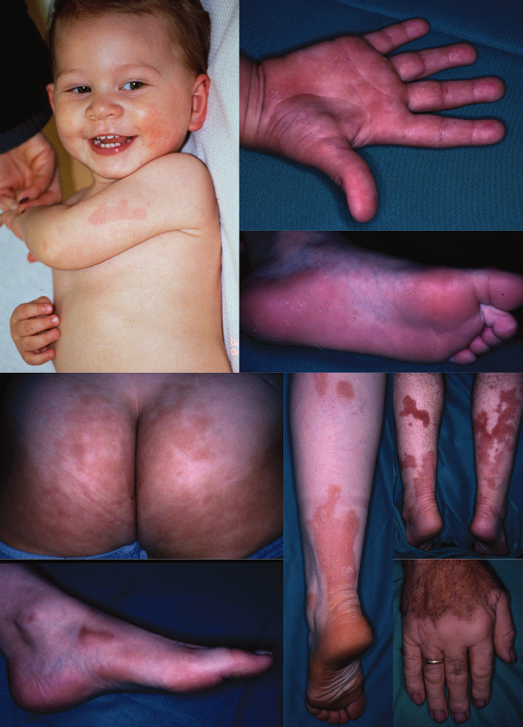
Prognosis.
Erythrokeratodermia variabilis normally continues into adulthood [38,41]. The hyperkeratotic plaques tend to appear or extend until puberty and then remain stable. In contrast, the erythematous lesions may decrease in intensity or even disappear after puberty [38,39].
Differential Diagnosis.
The transient and migratory nature of the erythematous lesions distinguishes EKV from other erythrokeratodermas and from pityriasis rubra pilaris, which is characterized by its folliculocentric involvement and an orange-coloured palmoplantar keratoderma. Older patients with EKV may present only with symmetric hyperkeratotic plaques such that differential diagnosis from progressive symmetric erythrokeratoderma is difficult without medical history or molecular genetic analysis. The differential diagnosis also includes non-classical variants of autosomal recessive congenital ichthyosis, Netherton syndrome, Greither palmoplantar keratoderma and keratolytic winter erythema (KWE; syn. erythrokeratolysis hiemalis). The latter two predominate on the palms and soles, and in KWE intermittent annular lesions with an exfoliative border can be seen on the distal limbs. Psoriasis and seborrhoeic dermatitis should be excluded by their classical clinical findings.
Treatment.
Before the development of the retinoids, a variety of therapies were tried, such as oral vitamin A, radiotherapy and intralesional steroids [42–45] with limited success and considerable side-effects such that they are not recommended. Historically, etretinate [46–48], and now acitretin [49,50] and isotretinoin have been shown to be effective treatments for both types of skin lesion of EKV. Emollients, topical retinoic acid and 5% lactic acid may be of some use in those intolerant of retinoids [34].
Erythrokeratodermia Variabilis Cram–Mevorah
Syn.
Annular erythrokeratodermia figurata variabilis, keratosis rubra figurata, erythrokeratodermia figurata variabilis type 2
History.
Cases of atypical EKV with distinctive circinate lesions were reported by Cram [37] and Hacham [51,52]. Identical cases may have been described as EKV of Degos, notably by Barrière [53]. Finally, similar families have been reported as ‘congenital ichthyosis with erythema centrifugum’ [54], ‘familial annular erythema’ [55] and ‘erythrokeratodermia anularis migrans’ [56].
Epidemiology.
About one-quarter of all EKV cases are due to mutations in Cx30.3 [28]. Of these at least one-quarter are characterized by circinate or erythema gyratum repens-like lesions [29]. Mevorah suggested that erythema repens-like lesions are characteristic of EKV cases with Cx30.3 mutations [52].
Pathogenesis.
Erythrokeratodermia variabilis Cram–Mevorah (EKV 2) is due to germline mutations (e.g. T85P or F137L) in the gap junction beta 4 gene (GJB4) that encode the gap junction protein connexin 30.3 (Cx30.3). On a molecular level, 30.3 mutations exert a dominant negative effect on connexin assembly with Cx31 [31].
Pathology.
Histology is non-specific and identical to EKV Mendes da Costa.
Clinical Features.
Skin lesions are present at birth or shortly thereafter. Migratory erythematous lesions tend to gradually turn into more or less fixed hyperkeratotic plaques. Some of these erythematous lesions appear as erythema gyratum repens and are characterized by rapidly migrating figurate erythema in an annular, garland or spiral arrangement (Fig. 122.3). Furthermore, slight peeling may be observed. Palms and soles are intact and audiograms are normal [52].
Fig 122.3 Erythrokeratoderma variabilis of the Cram–Mevorah type. Note the fractal like gyrate erythematous lesions.
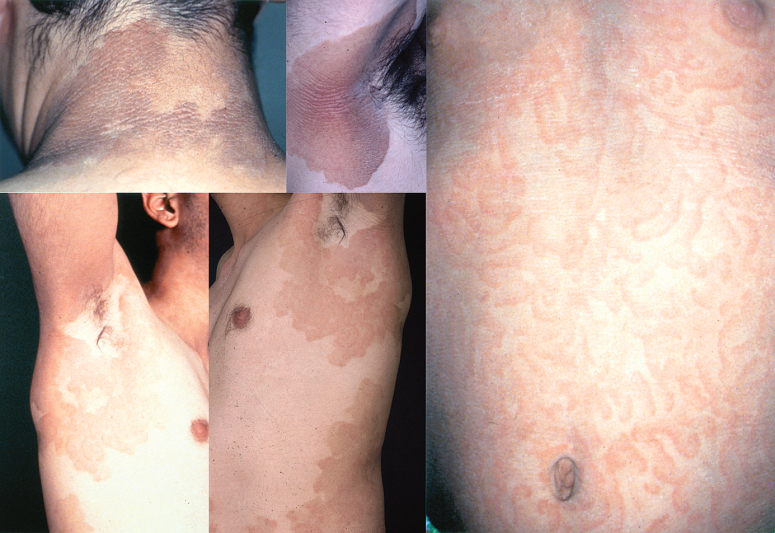
Prognosis.
Erythrokeratodermia variabilis Cram–Mevorah starts shortly after birth and continues into adulthood [37,52]. The hyperkeratotic plaques appear or extend until puberty and then remain stable. The erythematous lesions have a recurrent lifelong course. Nevertheless, EKV Cram–Mevorah tends to improve with age.
Differential Diagnosis.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


