*AD, autosomal dominant; AR, autosomal recessive; ASD, autosomal semi-dominant; ND, not determined; U, AR often heterozygous compound mutations in different genes; XD, X-linked dominant; XR, X-linked recessive.
Pathogenic Mechanisms in the MEDOC
Nature provides experiments. Indeed, the functional consequences of disturbed epidermal homeostasis and barrier formation are impressively demonstrated by the clinical phenotypes of numerous genodermatoses. The epidermal barrier constitutes a primordial frontline defence, which becomes established during terminal differentiation. This complex process involves proliferating basal keratinocytes becoming mitotically inactive and moving through four epidermal layers – the basal, spinous, granular and squamous layers. Throughout this differentiation programme, keratinocytes constantly modulate their proteomic profiles to the needs of the respective cell layer. As a result, stratum corneum cells contain polymerized bundles of keratin intermediate filaments and a water-retaining matrix surrounded by the cross-linked cornified cell envelope with ceramide lipids attached on the outer surface. These cells are concomitantly glued by intercellular lipid lamellae and are held together by corneodesmosmes.
During the long process of mammalian evolution, mechanisms to aid survival in the terrestrial environment have emerged. The surface of the Earth is affected by many quickly changing factors against which mammals need to be protected, such as ultraviolet (UV) radiation, wind, snow, injury and infection (pathogens). In order to deal with these challenges, the skin has to provide a barrier protecting the body against the environment. Its precise structure is designed to cope with mechanical, physical, chemical and microbial insults and water loss. The stratum corneum is the end-product of keratinocyte differentiation. Although composed of ‘dead cells’, it can be thought of as the ‘business end’ of the epidermis, providing the organism’s primary defensive barrier to the external environment.
A key function is the provision of a barrier to systemic evaporative water loss – a role that is essential for life in a dry, atmospheric environment. The permeability barrier is formed by lipid bilayers that fill the extracellular domain of the cornified layers. The lipids of the stratum corneum, which comprise ∼10% of its dry weight [8], are unusual in that the polar phospholipids, which are the predominant constituents of most cellular membranes, are absent. Instead, the stratum corneum membranes are composed of cholesterol, long-chain fatty acids and ceramides in approximately equimolar ratios [9,10]. This predominance of neutral lipids is ideally suited to the hydrophobic function of these membranes. These lipids form organized, repeating arrays of lamellae, which fill the intercellular spaces and, in conjunction with multiple corneocyte cell layers, form a tortuous, hydrophobic pathway that retards the outward movement of water [11]. Provision of sufficient lipid (quantity), both of the correct species and in the correct proportions (quality), is essential for formation of a competent permeability barrier. Stratum corneum lipids are derived largely from de novo synthesis in the epidermis. Therefore, the stratum corneum is particularly vulnerable to inborn errors of lipid metabolism. Several of the ichthyoses, including recessive X-linked ichthyosis, Sjögren–Larsson syndrome, the X-linked dominant ichthyoses (Conradi–Hunermann–Happle and CHILD syndromes), neonatal Gaucher and Niemann–Pick diseases, Refsum disease, neutral lipid storage disease (Chanarin–Dorfman syndrome) and some forms of autosomal recessive congenital ichthyosis (ARCI), are caused by lipid metabolic defects. In the latter, a specific novel lipid biosynthesis called the hepoxilin pathway has been identified [12]. Alterations in the lipid composition and structure of stratum corneum lamellar membranes have been identified in many of these lipidoses [13]. Lipids are delivered to the extracellular domain of the stratum corneum through secretion of a unique epidermal organelle, the lamellar body, at the junction of the stratum corneum and stratum granulosum.
In addition to lipids, a selected array of lysosomal enzymes is also packaged in lamellar bodies. Some of these are lipid hydrolases, required to convert glucosylceramide and phosphatidyl serine to ceramide, and to generate free fatty acids from other phospholipid precursors. Others are proteases, involved in the orderly degradation of corneodesmosomes that permit normal desquamation of corneocytes as single cells at the skin surface. Mutations affecting proteases and their inhibitors underlie Papillon–Lefèvre syndrome and Netherton syndrome respectively [14–16]. Also present in lamellar bodies are antimicrobial peptides, contributors to the innate immune defensive function of the stratum corneum [17,18]. The corneocytes provide a scaffold for the deposition of the intercellular membranes. This is achieved not only by the stacking of cell layers, but also through formation of the highly cross-linked protein cell boundary, the cornified cell envelope, to which some stratum corneum lipids are covalently attached to form the corneocyte-bound lipid envelope. Mutations affecting peptides of the cornified envelope (i.e. loricrin mutations in Camisa syndrome and variant progressive symmetric erythrokeratodermia) or the cross-linking enzyme, transglutaminase 1 (lamellar ichthyosis/congenital ichthyosiform erythroderma; CIE), are also causes of scaling disorders [19–24]. The cornified envelope also participates, along with the intracellular keratin intermediate filaments (KIF), in providing mechanical durability to the epidermis. Keratin intermediate filaments are the predominant subcellular structures of the corneocyte, after dissolution of the nucleus and other organelles. Mutations affecting keratins expressed in the outer epidermis, i.e. K1, K10 and K2E, underlie the generalized disorders epidermolytic ichthyosis (EI) and superficial epidermolytic ichthyosis (SEI; also known as ichthyosis bullosa of Siemens), whereas mutations affecting K9, K6a, K6b, K16 and K17 underlie the more localized palmoplantar keratodermas (PPK) of Vorner syndrome and pachyonychia congenita, respectively [25,26]. The expression pattern of the specific keratin gene/protein amongst and within different epithelia closely predicts the distribution of clinical involvement. Also present in corneocytes are small molecules derived from the hydrolysis of cellular contents, particularly the keratohyalin protein, profilaggrin. These small molecules provide an osmotic sink, to attract water into the corneocyte, and are required for normal stratum corneum hydration [27]. Moreover, the filaggrin breakdown products are acidic and contribute to the acidification of the stratum corneum, which in turn regulates the activity of several of the proteases involved in corneodesmosomal degradation (see below). Mutations resulting in deficiency of profilaggrin underlie ichthyosis vulgaris [28].
Corneodesmosomes provide the rivets between adjacent corneocytes. Gradual dissolution of these attachments by regulated proteolysis is essential to normal desquamation, and their absence leads to widespread blistering [29,30] while dominant-negative nonsense mutations lead to hypotrichosis simplex [31]. Defects in desmosomal constituents underlie several of the PPKs including striate PPK (desmoplakin, desmoglein I, cadherin), those with associated skin fragility (plakophilin) and with woolly hair (desmoplakin), or with woolly hair and cardiomyopathy (desmoplakin and plakoglobin) [32]. Loss of function of the serine protease inhibitor, LEKTI, which underlies Netherton syndrome [16], leads to accelerated digestion of corneodesmosomal attachments and, consequently, a thin stratum corneum [33]. The induction of TSLP by increased KLK5 activity resulting from the lack of inhibition by LETKI nicely exlains why atopic manifestations occur in Netherton syndrome [34]. Cholesterol sulphate is a protease inhibitor [35]. Accumulation of cholesterol sulphate in recessive X-linked ichthyosis stratum corneum as a result of steroid sulphatase deficiency results in impaired corneodesmosome degradation and retarded desquamation. Although the stratum corneum was viewed in the past as an inert tissue, it is instead an active regulator of epidermal homeostasis [8]. Acute insults to the barrier result in a sequence of signals to and responses from the underlying tissue aimed at restoring barrier function. These include: (i) secretion of preformed lamellar bodies; (ii) increased de novo epidermal lipid and peptide synthesis for the generation of more lamellar bodies; and (iii) stimulation of DNA synthesis. These messages – ‘Make more lipid and protease inhibitors! Make more cells!’ – are signalled through changes in the intercellular ionic milieu and through release of preformed pools of cytokines, particularly interleukin 1α (IL-1α), from the stratum corneum. These cytokines initiate a cytokine cascade that leads to epidermal hyperplasia and an inflammatory response. All of the ichthyoses studied to date have exhibited impaired barrier function; i.e. rates of transepidermal water loss (TEWL) are increased [36–38]. Although the stratum corneum is thicker than normal in most instances it is not ‘well made’ and functioning properly. Ultrastructural abnormalities in the integrity of the lamellar bilayers can be identified, even in disorders caused by mutations affecting non-lipid components [38]. The ichthyoses, therefore, present a situation analogous to experimental models of chronic barrier insufficiency (e.g. essential fatty acid deficiency), in which the barrier repair and proinflammatory signals cannot be extinguished because the underlying defect producing barrier dysfunction is unresolved. Hence, epidermal hyperplasia and some degree of inflammation (erythroderma) are common clinical features of many disorders of cornification (MEDOC). Epidermal hyperproliferation has its own consequences, including expression of alternative keratins, K6 and K16, and, when extreme, incomplete cornification, resulting in retained organelles (parakeratosis) and entombed lamellar bodies, all of which can be expected to further exacerbate the barrier insufficiency and drive the hyperplasia. Thus, the pathogenesis of the scaling phenotype is often likely to be more complex than a simple, linear consequence of perturbation in function of either the ‘bricks’ or the ‘mortar’. For example, scaling in recessive X-linked ichthyosis may be due to inhibition of corneodesmosome proteolysis rather than solely as a consequence of disturbance in the formation of the intercellular lipid lamellae by the accumulation of cholesterol sulphate [39]. Similarly, the blistering phenotype in epidermolytic ichthyosis is probably a direct consequence of the abnormal keratin intermediate filament network formed as the result of a dominant-negative mutation [40,41]. However, the scaling phenotype may also be caused by induction of particular signalling pathways [42], cytokines [41] and an impaired excretion of lamellar bodies secondary to derangement of the cytoskeleton, resulting in barrier insufficiency and the homeostatic responses described above [43]. Thus, in each DOC, complex and divergent pathogenic sequences may be initiated as the genetic defect impacts on normal stratum corneum formation and function.
Diagnostic Approaches
Although genetic testing is becoming increasingly available for these disorders, the multiplicity of genes implicated mandates a preliminary clinical diagnosis in order to direct testing towards the appropriate gene or genes. In establishing a clinical diagnosis, elements of both the history and the physical examination are critical. The age of onset, neonatal phenotype if congenital (Table 121.2), family history, including the presence of consanguinity, and the presence of any other medical problems are key historical data to be elicited. When examining the child, a systematic or algorithmic approach is helpful. Is the entire skin surface involved, or are there spared or skipped areas? Are the palms and soles involved; and if so, are they more, less or equally as severe as the rest of the body? Are flexures involved or spared? If involved, is the severity disproportionately severe or about the same as elsewhere? Is there facial involvement and is there ectropion? What is the quality of the scale pattern: light or dark, fine or thick and coarse (lamellar), or ridged? Is erythroderma present? Is the hair pattern normal?
Table 121.2 Neonatal presentations of MEDOC
| Neonatal presentation | Disorder |
| Collodion membrane | ARCI (approximately 70–80% collodion babies) |
| ‘Self-healing’ collodion baby | |
| Neonatal Gaucher disease | |
| Loricrin keratoderma | |
| Conradi–Hünermann–Happle syndrome (segmental) | |
| Netherton syndrome (occasionally) | |
| Ectodermal dysplasia | |
| KID syndrome (occasionally) | |
| Trichothiodystrophy (occasionally) | |
| Sjögren–Larsson syndrome (rarely) | |
| Neutral lipid storage disease (rarely)_ | |
| Massive constrictive scales | Harlequin ichthyosis |
| Neu–Laxova syndrome | |
| Erythroderma (with/without scale) | Netherton syndrome |
| ARCI | |
| KID syndrome | |
| CHIME syndrome | |
| Omenn syndrome | |
| Severe combined immunodeficiency (SCID) with maternal engraftment | |
| Hay–Wells syndrome | |
| X-linked hypohidrotic ectodermal dysplasia | |
| Scaling, hyperkeratosis or exaggerated neonatal desquamation (with/without erythema) | Recessive X-linked ichthyosis combined with heterozygous vulgar ichthyosis |
| Sjögren–Larsson syndrome | |
| Neutral lipid storage disorder | |
| Trichothiodystrophy | |
| Erosions/blisters, with/without erythema and hyperkeratosis | Epidermolytic ichthyosis |
| Superficial epidermolytic ichthyosis | |
| Excessive ‘vernix’ | Ichthyosis prematurity syndrome |
| KID syndrome | |
| Harlequin ichthyosis | |
| Keratoses and erythroderma in Blaschkoid distribution | Conradi–Hünermann–Happle syndrome |
| Incontinentia pigmenti | |
| Inflammatory linear verrucous epidermal naevus (ILVEN) | |
| Transient erythematous patches, irregularly shaped | Erythrokeratoderma variabilis |
| Normal skin | Refsum disease |
| Neutral lipid storage disorder | |
| Ichthyosis vulgaris | |
| Recessive X-linked ichthyosis | |
| Multiple sulphatase deficiency | |
| Superficial epidermolytic ichthyosis | |
| Erythrokeratodermia variabilis | |
| Erythrokeratodermia progressiva symmetrica | |
| Netherton syndrome |
ARCI, autosomal recessive congenital ichthyosis; CHIME, colobomas, congenital heart disease, early-onset ichthyosiform dermatosis, mental retardation and ear abnormalities; KID, keratitis, ichthyosis and deafness.
Once a clinical diagnosis is achieved, diverse laboratory approaches often allow its confirmation. Skin biopsy is rarely diagnostic, although there are notable exceptions, such as ichthyosis vulgaris, epidermolytic hyperkeratosis (EI), loricrin keratoderma or Conradi–Hünermann–Happle syndrome. Immunohistochemistry is increasingly applied, notably in autosomal recessive disorders due to protein deficiencies, for example congenital ichthyoses, IHSC (see Table 121.1) or Netherton syndrome. Electron microscopy is helpful only exceptionally and requires an interpreter who is experienced in evaluating these rare skin disorders. Molecular genetic testing is of great value. However, the clinician should bear in mind that genetic testing is not always confirmative, even when the clinical diagnosis is correct. Sometimes this occurs because more than one gene can cause a clinical disorder. In EI phenotypes or in lamellar ichthyoses, for example, it may be necessary to screen for the keratin genes KRT1 and KRT10 or for different enzyme deficiencies, respectively. Furthermore, the genetic test employed may not be broad or inclusive enough to detect the patient’s mutation. Most laboratories only screen for ‘hotspot’ mutations; thus, an uncommon mutation lying outside these regions will not be detected. Similarly, in recessive X-linked ichthyosis, fluorescent in situ hybridization (FISH) or polymerase chain reaction (PCR) tests only detect large or complete deletions of the steroid sulphatase gene. Small deletions or point mutations will be read as ‘normal’. Therefore, in cases where the clinical diagnosis is well founded, negative genetic tests should not be overinterpreted and, when possible, should be further evaluated, such as by measurement of enzyme activity in recessive X-linked ichthyosis (RXLI). Finally, even with exhaustive searching, some patients with distinct phenotypes will not exhibit identifiable gene mutations. This suggests that other as yet unidentified genes can produce similar phenotypes, or perhaps regulators of the gene’s transcription or translation are affected. The failure of genetic confirmation does not necessarily negate an otherwise well-founded clinical diagnosis. Nonetheless, confirmatory genetic testing provides the highest level of certainty to diagnosis. Moreover, when the specific mutation in a family is known, genetic screening for carrier status and for affected family members is greatly facilitated, especially with the time constraints of prenatal diagnosis during early pregnancy.
Taking a lead from the recent consensus conference, the MEDOC are considered under the following four headings: Common ichthyoses and related syndromes, Keratodermic ichthyoses, Autosomal recessive congenital ichthyoses and Syndromic ichthyoses
References
1 Riecke E. Über Ichthyiosis congenita. Archiv fuer Dermatologie und Syphilis 1900;54:289–340.
2 Brocq, L. Erythrodermie congenitale ichthyosiforme avec hyperepidermotrophie. Ann Dermatol Syph 1902;4:1–31.
3 Siemens, H. Dichtung und Wahrheit über die Ichthyosis bullosa, mit Bemerkungen zur Systematik der Epidermolysen. Arch Dermatol Syph 1937;175:590–608.
4 Wells RS, Kerr CB. Clinical features of automal dominant and sex-linked ichthyosis in an English population. Brit Med J 1966;1:947–50.
5 Weibel ER, Schnyder UW. [On the ultrastructure and histochemistry of granular degeneration in vesicular ichthyosiform congenital erythrodermia]. Arch Klin Exp Dermatol 1966;227:341–2.
6 Frost P, Van Scott EJ. Ichthyosiform dermatoses. Classification based on anatomic and biometric observations. Arch Dermatol 1966;94:113–26.
7 Oji V, Tadini G, Traupe H, et al. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol 2010.
8 Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res 2008;49:697–714.
9 Elias PM, Williams ML, Maloney ME et al. Stratum corneum lipids in disorders of cornification. Steroid sulfatase and cholesterol sulfate in normal desquamation and the pathogenesis of recessive X-linked ichthyosis. J Clin Invest 1984;74:1414–21.
10 Wertz PW. Epidermal lipids. Semin Dermatol 1992;11:106–13.
11 Potts RO, Francoeur ML. The influence of stratum corneum morphology on water permeability. J Invest Dermatol 1991;96:495–9.
12 Yu Z, Schneider C, Boeglin WE, Brash AR. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARalpha. Lipids 2007;42:491–7.
13 Schmuth M, Man MQ, Weber F et al. Permeability barrier disorder in Niemann–Pick disease: sphingomyelin-ceramide processing required for normal barrier homeostasis. J Invest Dermatol 2000;115:459–66.
14 Toomes C, James J, Wood AJ et al. Loss-of-function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet 1999;23:421–4.
15 Hart TC, Hart PS, Bowden DW et al. Mutations of the cathepsin C gene are responsible for Papillon–Lefevre syndrome. J Med Genet 1999;36:881–7.
16 Chavanas S, Bodemer C, Rochat A et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet 2000;25:141–2.
17 Braff MH, Di Nardo A, Gallo RL. Keratinocytes store the antimicrobial peptide cathelicidin in lamellar bodies. J Invest Dermatol 2005;124:394–400.
18 Schauber J, Gallo RL. Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol 2009;124:R13–18.
19 Maestrini E, Monaco AP, McGrath JA et al. A molecular defect in loricrin, the major component of the cornified cell envelope, underlies Vohwinkel’s syndrome. Nat Genet 1996;13:70–7.
20 Ishida-Yamamoto A, Takahashi H, Iizuka H. Loricrin and human skin diseases: molecular basis of loricrin keratodermas. Histol Histopathol 1998;13:819–26.
21 Suga Y, Jarnik M, Attar PS et al. Transgenic mice expressing a mutant form of loricrin reveal the molecular basis of the skin diseases, Vohwinkel syndrome and progressive symmetric erythrokeratoderma. J Cell Biol 2000;151:401–12.
22 Schmuth M, Fluhr JW, Crumrine DC. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis). J Invest Dermatol 2004;122:909–22.
23 Huber M, Rettler I, Bernasconi K et al. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science 1995;267:525–8.
24 Russell LJ, DiGiovanna JJ, Rogers GR et al. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet 1995;9:279–83.
25 Smith F. The molecular genetics of keratin disorders. Am J Clin Dermatol 2003;4:347–64.
26 McLean WH, Irvine AD. Disorders of keratinisation: from rare to common genetic diseases of skin and other epithelial tissues. Ulster Med J 2007;76:72–82.
27 Rawlings AV, Scott IR, Harding CR, Bowser PA. Stratum corneum moisturization at the molecular level. J Invest Dermatol 1994;103:731–41.
28 Smith FJ, Irvine AD, Terron-Kwiatkowski A et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 2006;38:337–42.
29 Matsumoto M, Zhou Y, Matsuo S et al. Targeted deletion of the murine corneodesmosin gene delineates its essential role in skin and hair physiology. Proc Natl Acad Sci USA 2008;105:6720–4.
30 Leclerc EA, Huchenq A, Mattiuzzo NR et al. Corneodesmosin gene ablation induces lethal skin-barrier disruption and hair-follicle degeneration related to desmosome dysfunction. J Cell Sci 2009;122:2699–709.
31 Levy-Nissenbaum E, Betz RC, Frydman M et al. Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet 2003;34:151–3.
32 Lai-Cheong JE, Arita K, McGrath JA. Genetic diseases of junctions. J Invest Dermatol 2007;127:2713–25.
33 Deraison C, Bonnart C, Lopez F et al. LEKTI fragments specifically inhibit KLK5, KLK7, and KLK14 and control desquamation through a pH-dependent interaction. Mol Biol Cell 2007;18:3607–19.
34 Briot A, Deraison C, Lacroix M et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med 2009;206:1135–47.
35 Elias PM, Crumrine D, Rassner U et al. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol 2004;122:314–19.
36 Frost P, Weinstein GD, Bothwell JW, Wildnauer R. Ichthyosiform dermatoses. 3. Studies of transepidermal water loss. Arch Dermatol 1968;98:230–3.
37 Lavrijsen AP, Oestmann E, Hermans J, Bodde HE, Vermeer BJ, Ponec M. Barrier function parameters in various keratinization disorders: transepidermal water loss and vascular response to hexyl nicotinate. Br J Dermatol 1993;129:547–53.
38 Moskowitz DG, Fowler AJ, Heyman MB et al. Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr 2004;145:82–92.
39 Zettersten E, Man MQ, Sato J et al. Recessive X-linked ichthyosis: role of cholesterol-sulfate accumulation in the barrier abnormality. J Invest Dermatol 1998;111:784–90.
40 Reichelt J, Magin TM. Hyperproliferation, induction of c-Myc and 14-3-3sigma, but no cell fragility in keratin-10-null mice. J Cell Sci 2002;115:2639–50.
41 Roth W, Reuter U, Wohlenberg C, Bruckner-Tuderman L, Magin TM. Cytokines as genetic modifiers in K5–/– mice and in human epidermolysis bullosa simplex. Hum Mutat 2009;30:832–41.
42 Vijayaraj P, Kroger C, Reuter U, Windoffer R, Leube RE, Magin TM. Keratins regulate protein biosynthesis through localization of GLUT1 and -3 upstream of AMP kinase and Raptor. J Cell Biol 2009;187:175–84.
43 Schmuth M, Yosipovitch G, Williams ML et al. Pathogenesis of the permeability barrier abnormality in epidermolytic hyperkeratosis. J Invest Dermatol 2001;117:837–47.
Common Ichthyoses and Related Syndromes
Ichthyosis Vulgaris
History and Epidemiology.
Ichthyosis vulgaris (OMIM 146700, IV) is the most common and mildest form of generalized ichthyosis. Although ‘common’ forms of ichthyosis were recognized and well described by the end of the 19th century by Riecke, the large, population-based studies of Wells and Kerr [1] in the mid-20th century defined an autosomal dominant mode of transmission with a frequency of 1 : 250, distinguishing ichthyosis vulgaris from the less common recessive X-linked ichthyosis (RXLI). The true semi-dominant mode of transmission of ichthyosis vulgaris became apparent by the identification of its recessive transmission as flaky tail trait in mice [2] and the subsequent groundbreaking work of McLean, Irvine and collaborators identifying profilaggrin mutations as the bona fide cause of the human disease [3]. Ichthyosis vulgaris exists in two forms. The classic form with a more severe phenotype is due to biallelic profilaggrin mutations with a frequency of 1/700 and thus, surprisingly, ichthyosis vulgaris is transmitted in a semi-dominant fashion. The more common single allele or heterozygous form occurs in about 10% of the European population. This heterozygous ichthyosis vulgaris shows significant clinical overlap with xerosis, i.e., with the dry skin that accompanies atopic dermatitis. Indeed profilaggrin mutations are the major risk factor for atopic dermatitis [4,5]. The severity of the heterozygous form is more variable, particularly in response to regional and seasonal changes in temperature and humidity. It seems likely that some of this variability may also be due to the influence of other modifier genes, such as, perhaps, heterozygosity for mutations in other ichthyosis genes.
Clinical Features.
Although classic ichthyosis vulgaris is not present at birth, physical signs of the condition usually manifest within the first year of life, with onset by 3 months of age in 40% of affected individuals [6,7]. Due to the semi-dominant transmission of ichthyosis vulgaris the clinical expression varies from minor heterozygous forms primarily located on palmoplantar and extensor surfaces of the legs, to complete biallelic forms with a more generalized extension. This classic ichthyosis vulgaris is characterized by prominent scaling with small, fine, white to grey scales, which are often larger and more adherent on the extensor surfaces. Thus, the extremities are generally more severely affected than the torso. Facial involvement is uncommon and characterized by a predilection for the forehead and cheeks; heterozygous forms have only dry facial skin and accentuated lines of the lips. Flexural areas are spared, and this remains an important finding for clinical separation from other types of ichthyoses. The greatest area of uninvolved skin occurs at large flexures, such as the axillae. The clinical severity varies, depending on the temperature and ambient humidity; scaling is often improved in warm, humid environments.
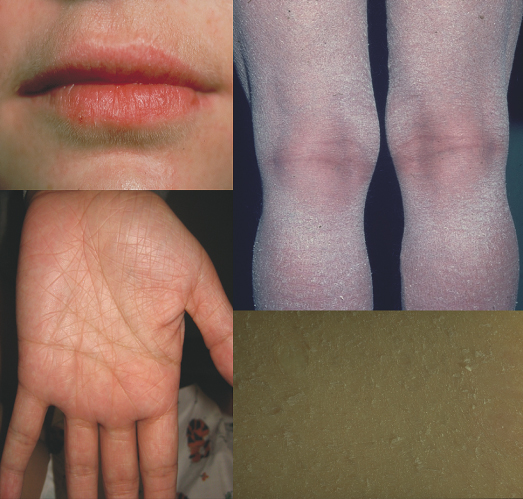
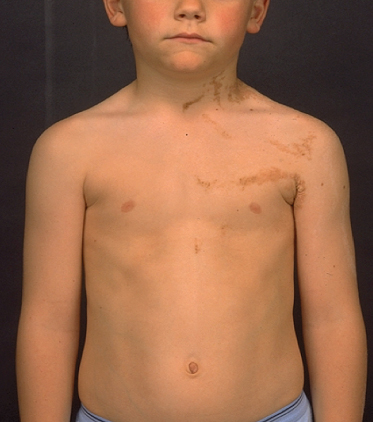
Other cutaneous findings associated regularly and also observed in heterozygous ichthyosis vulgaris include keratosis pilaris, and hyperlinear palms and soles or lips (Parrot sign). Keratosis pilaris is seen in up to 75% of patients with ichthyosis vulgaris, compared with 40% of control subjects [8]. Mevorah noted early that hyperlinear palms and soles characterized by accentuated creases (Fig. 121.1) are a most reliable sign of ichthyosis vulgaris, and recent molecular work confirmed that they reflect best the degree of profilaggrin deficiency [3]. These changes have also been reported as minor diagnostic features of atopic dermatitis [8,9] since about 35–50% of all atopic patients suffer from profilaggrin deficiency [4]. Conversely, atopic dermatitis is present in over half of ichthyosis vulgaris cases. Keratosis punctata of the palmar creases may be associated with ichthyosis vulgaris. Other than its association with atopy, ichthyosis vulgaris lacks extracutaneous findings.
Fig. 121.1 Hyperlinear palms, soles and lips, an obligate but non-specific feature of ichthyosis vulgaris.
Histological and Ultrastructural Features.
The key histological features of ichthyosis vulgaris are a retention hyperkeratosis with a normal epidermis covered by a compact orthokeratotic hyperkeratosis. The granular cell layer is diminished or absent [10,11]. The expression of the granular layer, however, varies in heterozygous ichthyosis vulgaris from individual to individual and from one body site to another. Biallelic ichthyosis vulgaris shows histological absence of the granular layer as a constant finding on multiple body sites over time, and this correlates with the lack of keratohyalin granules on histology and electron microscopy [12]. Light microscopy is a convenient means of diagnosing ichthyosis vulgaris. Electron microscopy studies demonstrate absent or reduced numbers of keratohyalin granules [13,14]. The morphology of keratohyalin granules has been described as abnormal (‘crumbly’) [13] but this appears to arise from an artefact of fixation [12].
Genetics and Pathogenesis.
Early biochemical work revealed that deficiencies of the precursor protein profilaggrin in the stratum granulosum and its product filaggrin (filament-aggregating protein) in the stratum corneum are associated with ichthyosis vulgaris, and that profilaggrin deficiency correlates with reduced or absent keratohyalin granules morphologically and with disease severity clinically [14,15]. Profilaggrin is encoded by the large FLG gene located in the epidermal differentiation complex on chromosome 1q21 and is one of the key proteins of the epidermis. Together with loricrin, profilaggrin accounts for about 75% of the entire transcriptional protein product of the granular layer. The initial product of the FLG gene is the large profilaggrin protein of ∼435 kDa with a domain structure: an N-terminus S100 calcium-binding domain consisting of two EF-hands, corresponding to the A domain; a B domain with a nuclear localization signal, 10 to 12 allelic filaggrin peptide repeats flanked both sides by imperfect filaggrin repeats; and a C-terminal domain.
Profilaggrin has a high content of phosphorylated histidine and is the main constituent of keratohyalin granules. As granular cells undergo late terminal differentiation to form corneocytes, profilaggrin is dephosphorylated and as a consequence proteolytically cleaved by caspase-14 into the multiple repeated filaggrin peptides. Filaggrin binds to and aggregates the keratin cytoskeleton in the lower stratum corneum. In the upper stratum corneum filaggrin is mostly degraded to free amino acids, urocanic acid and other small acidic molecules, which have the capacity to bind water [16]. Filaggrin hydrolysis is regulated by external humidity and is triggered by xeric stress [17]. In addition to stratum corneum hydration, these breakdown products play important roles in acidification, flexibility and the UV-barrier [18].
Reduced amounts of profilaggrin and filaggrin are expressed in the epidermis of the Flaky Tail mouse, a model for human ichthyosis vulgaris with autosomal recessive transmission [2]. Ichthyosis vulgaris is inherited as an autosomal semi-dominant trait and has been shown to be due to mutations of the FLG gene [3], confirming linkage analysis by Compton et al. [19]. Homozygous FLG mutations lead to complete loss of profilaggrin expression in skin, and the absence of hygroscopic molecules critical to maintaining epidermal hydration translates clinically to the dry and scaly skin characteristic of the disease. About 1 in 700 Europeans suffers from homozygous ichthyosis vulgaris due to transmission of two profilaggrin gene null alleles, and about 1 in 10 Europeans has heterozygous profilaggrin deficiency. The latter is transmitted as a semi-dominant trait now identified as heterozygous ichthyosis vulgaris. A number of recurrent mutations have been identified in this population, while other recurrent filaggrin mutations are prevalent in Chinese and in Japanese populations [20–22]. Ichthyosis vulgaris is the major susceptibility factor predisposing for atopic dermatitis, with overwhelming statistical evidence (p-value 10−17), and to a lesser extent for asthma [4,23]. In fact, FLG mutations can be identified in 50% of children with atopic dermatitis [4].
The pathogenesis of scaling in ichthyosis vulgaris is complex. In addition to deficiency of filaggrin-derived small molecules required for corneocyte hydration, stratum corneum acidification may be impaired. Moreover, preliminary studies in genotyped ichthyosis vulgaris subjects suggest that the phenotype in ichthyosis vulgaris is linked to an underlying abnormality in the permeability barrier [24]. Although filaggrin monomers are believed to mediate the collapse of the keratin filament network during cornification, keratins appear to have a normal ultrastructural appearance in the cytosol of ichthyosis vulgaris corneocytes [14,15], suggesting that filaggrin may not be required for normal keratin aggregation.
Differential Diagnosis.
As indicated, the clinical phenotype of ichthyosis vulgaris merges with that of severe xerosis, and accounts in large part for the xerosis associated with atopic dermatitis. Flexural sparing is a key clinical feature of ichthyosis vulgaris that helps differentiate it from most other ichthyoses, with the exception of recessive X-linked ichthyosis (RXLI) (Fig. 121.2). If flexures are involved, a mild lamellar ichthyosis/congenital ichthyosiform erythroderma (LI/CIE) phenotype is a more likely diagnosis. In males, clinical distinction from RXLI may not be possible (see Table 121.3), and biochemical or genetic testing for RXLI may be needed to secure a diagnosis. In those cases with disproportionate palmoplantar hyperkeratosis, loricrin keratoderma (Vohwinkel syndrome, Camisa type) should be considered. Associated early-onset, progressive mental and neurological impairment suggests multiple sulphatase deficiency.
Table 121.3 Clinical and histopathological features of ichthyosis vulgaris and recessive X-linked ichthyosis (RXLI)
| Feature | Ichthyosis vulgaris | RXLI |
| Age of onset | 3–6 months | Birth to 3 weeks |
| Scale | Small, white to grey, flaky | Small to large, brownish, polygonal, tightly adherent |
| Characteristic distribution: | ||
| Face (if involved) | Usually spared (forehead, cheeks) | Preauricular |
| Neck | Spared | Mild to moderate |
| Torso | Mild | Moderate |
| Extremities | Mild to moderate | Moderate |
| Palms, soles and lips | Hyperlinear with accentuated creases | Spared |
| Flexural involvement | Marked sparing, all flexures | Antecubital and popliteal spared; neck and axillae involved |
| Histology: hyperkeratosis | Compact orthohyperkeratosis | Compact orthohyperkeratosis |
| Histology: granular layer | Reduced if heterozygous Absent if biallelic | Normal |
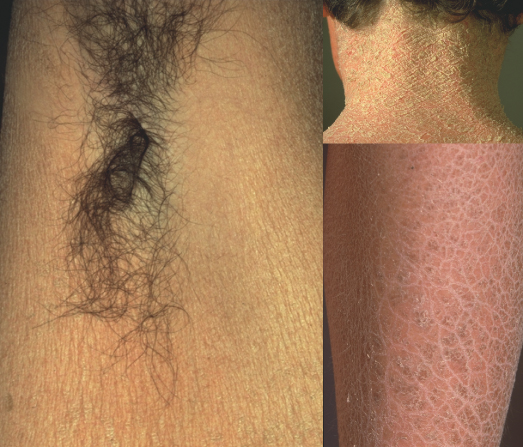
Fig. 121.2 Flexural sparing in RXLI. Antecubital fossae and popliteal fossae are always spared to some degree. This feature can differentiate between RXLI and a mild ARCI phenotype, but does not distinguish RXLI from IV. The presence of a dark colour of the scale and the absence of hyperlinearity and follicular keratosis favours RXLI.
Dry, hyperkeratotic skin with keratosis pilaris is also seen in the cardiofaciocutaneous syndrome (CFC, OMIM 115150), a rare disorder characterized by congenital heart defects, distinct facies, short stature, mental retardation and ectodermal abnormalities [25]. Additional skin changes include eczematous patches and loose skin on the hands and feet. Hair is typically sparse, slow growing and curly, and eyebrows and eyelashes are decreased to absent. Nails are slow growing as well. Cardiofaciocutaneous syndrome shows considerable phenotypic overlap with Noonan (OMIM 163950, PTPN11) and Costello (OMIM 218040, HRAS) syndromes, but mutations of KRAS, BRAF, MEK1 or MEK2 have been identified as the underlying cause. The protein products of these genes interact in a common RAS/ERK pathway that regulates cell differentiation, proliferation and apoptosis [26].
Finally, as ichthyosis vulgaris usually presents in infancy, alternative diagnoses should be considered in older children or adolescents who develop ichthyosis vulgaris-like skin changes. Associated late-onset visual and neurological abnormalities suggest a diagnosis of Refsum disease. Acquired ichthyosis is generally considered a disorder of adults, but appears clinically indistinct from hereditary ichthyosis vulgaris [27]. It is a cutaneous marker for many underlying disorders (Table 121.4), and evaluation should be directed by a thorough history and review of systems.
Table 121.4 Xp contiguous gene deletions associated with recessive X-linked ichthyosis (RXLI)
| Gene(s)* (symbol) | Disorder | Clinical features |
| Short stature homeobox gene (SHOX) | Idiopathic familial short stature | Short stature |
| Arylsulphatase E (ARSE) | X-linked recessive chondrodysplasia punctata | Nasal hypoplasia, distal phalangeal hypoplasia, short limbs, ‘stippled’ cartilage |
| Variably charged X (VCX3A) and other obligatory genes | X-linked mental retardation | Limited cognitive function |
| Neuroligin 4 (NLGN4) | X-linked autism, ADHD | Autism, social communication difficulties, hyperactivity |
| Steroid sulphatase (STS) | RXLI | Ichthyosis, placental sulphatase deficiency |
| Kallmann syndrome-1, anosmin-1 (KAL1) | Kallmann syndrome | Hypogonadotropic gonadism, anosmia/hyposmia, renal anomalies, obesity, synkinesia, high arched palate, cerebellar ataxia |
| Ocular albinism-1, Nettleship–Falls type (OA1) | Ocular albinism, Nettleship–Falls type | Nystagmus, decreased visual acuity, macular hypoplasia, ocular hypopigmentation |
*Genes are listed in order from proximal to distal on Xp. Multiple nomenclatures are indicated.
ADHD, attention deficit hyperactivity disorder.
Management.
Ichthyosis vulgaris is best controlled with the aggressive use of emollients combined with a practical approach to skin care that is non-irritating and does not exacerbate xerosis. Bathing should occur only as frequently as needed and should be limited to a short period of time with tepid water. Synthetic detergents, rather than soaps, should be used. Within 5 minutes of bathing, emollients should be applied. Reapplication when the skin is dry is also indicated. Products containing humectants and/or keratolytics may provide additional benefit if conservative measures are not adequate. Some products, however, may produce a burning sensation, especially in those with concurrent atopic dermatitis. Thus, aqueous preparations such as lotions or creams containing 5% or less urea or lactic acid are often preferred over more aggressive keratolytic ointments. The use of local retinoids or vitamin D analogues and systemic retinoids for ichthyosis vulgaris are not indicated and are particularly prone to reveal or exacerbate atopic dermatitis.
Recessive X-Linked Ichthyosis
History and Epidemiology.
Recessive X-linked ichthyosis, a deficiency of the enzyme steroid sulphatase (STS) (RXLI, OMIM 308100), was the first genodermatosis to be molecularly defined [28]. This relatively common and well-described disorder occurs worldwide with a frequency that ranges from 1 in 2000 to 10,000 males, depending on the study population and method of assessment [6,29–31]. At least since the thesis of Riecke in 1900 it had been recognized that a non-congenital or vulgar form of ichthyosis primarily affected males. However, only the population-based studies of Wells and Kerr [6] clearly separated RXLI from ichthyosis vulgaris as a clinical entity based on clinical criteria and its mode of inheritance. Concurrently, France and Liggins [32] linked the absence of STS in placentas of some male fetuses to abnormal maternal oestrogen profiles. Termed ‘placental sulphatase deficiency syndrome’, the disorder was characterized by failure of labour initiation or progression, but delivery of an otherwise well, male infant. Jobsis and colleagues first suggested the relationship of STS deficiency to X-linked ichthyosis. Soon after, the studies of Koppe and colleagues [28] and Shapiro and colleagues [33] documented deficient STS activity in skin fibroblasts from patients with RXLI.
Clinical Features.
Although the clinical features of RXLI can range from mild to marked and may overlap with those of ichthyosis vulgaris, an accurate clinical diagnosis can often be made by careful observation (see Table 121.3). Absence of flexural sparing, palmoplantar hyperlinearity, follicular keratoses and atopy are features in favour of RXLI. Recessive X-linked ichthyosis commonly presents within the first weeks of life as exaggerated desquamation [34]. In case of concurrent heterozygous vulgar ichthyosis it may be present at birth (D. Hohl, unpublished observation) and be difficult to separate from mild autosomal recessive congenital ichthyosis. Over the next few months, prominent scale that is characteristically large, brown, geometrically shaped and often tightly adherent replaces normal skin. It is symmetrically distributed over the torso and extremities, and most pronounced over the extensor surfaces, especially the legs [7,34]. The lateral and posterior aspects of the neck are often covered with dark scales giving a ‘dirty’ appearance, previously described as ‘ichthyosis nigricans’ (see Fig. 121.2) [6]. Scaling often encroaches on flexural surfaces; however, the antecubital and popliteal fossae will be spared to some extent in RXLI [7,34,35]. The preauricular region is another characteristic site of involvement, although the remainder of the face is usually spared [35]. The scalp is almost always affected in childhood, but usually improves over time, in contrast with the trunk and extremities, where scale is persistent [34]. The palms and soles are usually unaffected in RXLI, and hair and nails are normal [7,34,35]. Similar to ichthyosis vulgaris, most patients with RXLI improve during the warmer and more humid summer months.
Extracutaneous Features.
Although commonly considered one of the ‘primary’ or ‘non-syndromic’ forms of ichthyosis, RXLI is a systemic disorder that may be associated with extracutaneous signs. The first extracutaneous signs, ironically, are apparent in pregnant females carrying affected male fetuses. The placenta, a predominantly fetal structure, plays a major role in the production of oestriol (unconjugated estriol, uE3) during pregnancy. Lack of STS in affected males leads to low levels of maternal unconjugated oestriol [36], which may be noted on routine screening offered between 15 and 20 weeks of gestation. Although an isolated low level of uE3 is a sensitive marker for RXLI, it is not specific, as decreased uE3 can be seen in a variety of other conditions such as fetal demise, trisomy 18 and 21, neural tube defects and Smith–Lemli–Opitz syndrome [37,38]. Parturition in placental STS deficiency may be complicated by delayed onset of labour, failed cervical softening and dilation, and a suboptimal response to psilocin [1,11], which may lead to forceps/vacuum-assisted delivery or caesarean section. Infants with RXLI are generally healthy after birth.
Corneal opacities can be found in approximately 10–50% of patients and female carriers [39,40]. These diffuse corneal deposits are located in the posterior capsule of Descement’s membrane or corneal stroma and do not impair visual acuity. Although corneal opacities may be present at any age, some authors have found them to be more frequent during the second to third decades of life [41,42].
A number of extracutaneous abnormalities have been reported in males with RXLI. Many of these abnormalities have been shown to be part of ‘contiguous gene syndromes’ (see below), arising as a result of deletions of genes neighbouring the STS gene locus. Other reports, however, suggest that STS deficiency may have other systemic associations. Testicular maldescent (most often inguinal cryptorchidism) occurs more frequently in males with RXLI. Normally seen in about 1% of males, it is reported to occur in approximately 12–28% of those affected with RXLI [43]. Steroid sulphatase is expressed in the testes, but its role in testicular descent remains unclear [43]. Testicular cancer was reported in two men with RXLI, both of whom had normally descended testes [44]. A lack of further reports, however, suggests this may be a coincidental association. The coexistence of RXLI and hypertrophic pyloric stenosis has been reported in two families: one with three affected brothers and the other with affected dizygotic male twins [45,46]. Other likely coincidental associations reported with RXLI include an infant born with exstrophia cloacae and omphalocele, renal failure [47], bilateral periventricular nodular heterotopias [48] and a case of acute lymphoblastic leukaemia [49]. Others have suggested an increased risk of male pattern baldness in RXLI.
Finallly, there is convincing evidence that Xp deletions and RXLI cause cognitive behavioural features such as attention deficit hyperactivity disorder (ADHD) [50], mental retardation [51,52] and autism [53], presumably on the basis of deletion of neighbouring genes contiguous to STS (see below). However, recent reports suggest that STS deficiency per se is a risk factor for ADHD, predominantly with inattentive symptoms [54].
Contiguous Gene Syndromes.
In the past, the uncommon association of RXLI with other rare disorders such as hypogonadotropic hypogonadism or mental retardation was thought to represent a novel genetic syndrome, ‘Rud syndrome’ [55–57]. More recently, it has become apparent that these associations arise as a result of deletions of genes residing near the STS gene locus [58,59]. Deletion of the entire STS gene, rather than point mutations or smaller deletions, is the cause of the vast majority of RXLI cases (see below). Concomitant deletion of neighbouring loci results in a contiguous gene syndrome (CGS). The phenotypes in these syndromes are best thought of as concurrences of two or more monogenic disorders.
The prevalence of CGS associated with RXLI from a referral centre was as high as 25% [60]. Our own unpublished series of more than 50 cases indicates that it occurs less frequently, at most in 5% of cases. The most commonly associated disorder is Kallmann syndrome, an uncommon disorder characterized by hypogonadotropic hypogonadism and anosmia, which is due to deletions of the KAL1 gene encoding anosmin. Other less frequent features of Kallmann syndrome include renal agenesis, mirror movements of the hands (bimanual synkinesia), pes cavus, high arched palate and cerebellar ataxia [61]. The finding of cryptorchidism in males with RXLI should alert the clinician to the possibility of coexistent Kallmann syndrome. In such cases, prompt institution of hormone replacement can prevent complications such as delayed puberty, infertility, obesity and osteoporosis [60,62]).
Other associated deletions include X-linked chondrodysplasia punctata (ARSE), short stature (SHOX), X-linked mental retardation and autism [54,59,63]. Indeed, several case reports of patients with Xp deletions and RXLI include cognitive behavioural features such as ADHD [50], mental retardation [51,52] and autism [53]. The VCX3A gene (variably charged, X chromosome, 3A; also known as VCX-A) may have a role in the occurrence of X-linked mental retardation (XLMR) in some RXLI individuals [51]; however, deletion of VCX3A alone is not sufficient [64]. Transmission of XLMR appears highly complex and involves at least nine genes [65]. The neuroligin 4 (NLGN4) gene, which is included in the deleted region in some RXLI patients, may have a role in the autism and social communication difficulties since mutations in this gene have been associated with autism [66]. Further, cases of RXLI with large deletions encompassing STS and NLGN4 are at increased risk of developing autism [67].
Ocular albinism of the Nettleship–Falls type (OA1) has been reported in a family with documented RXLI, but without Kallmann syndrome. The OA1 gene has been mapped to the Xp22.3 to Xp22.2 region, but is proximal to the STS and Kallmann genes [60], suggesting that the reported coexistence of RXLI and OA1 is either coincidental or the result of a complex genetic rearrangement and deletion.
Histological and Ultrastructural Features.
The characteristic histological features of RXLI are compact hyperkeratosis of the stratum corneum and a normal or slightly thickened granular layer [7,11,68]. Unfortunately, these findings are non-specific. Acanthosis, papillomatosis, focal parakeratosis and follicular plugging may be seen on occasion as well [7,11].
Early ultrastructural observation in four cases of RXLI [69] revealed increased keratohyalin granules, persistent corneodesmosomes in the outermost cornfield layers and corneocytes containing large numbers of melanosomes, possibly accounting for the dark scale. Elias and collaborators observed in addition abnormal stratum corneum lamellar membrane structures with formation of structureless lacunae [70].
Genetics and Cutaneous Pathogenesis.
Recessive X-linked ichthyosis is inherited as an X-linked recessive trait; heterozygous females are clinically unaffected. Affected, homozygous females are reported, albeit rarely [71,72]. The STS gene locus is localized to the distal portion of the short arm of the X chromosome (Xp22.3) [58,73,74], a region prone to deletions due to its high frequency of unequal crossovers during meiosis. More than 90% of cases arise due to complete deletion of the STS gene [75,76], making RXLI the genetic disorder with the greatest incidence of complete gene deletions [75]. The regional tendency towards genomic deletions also produces RXLI-associated CGS (see above). In a minority of cases, RXLI is the result of partial gene deletions or point mutations [77]. The cutaneous phenotypes in these two groups of patients are similar.
The product of the STS gene, STS, is a hydrolytic microsomal enzyme that cleaves the sulphate group at the 3β position from sterols and steroids (for a recent review, see Ref. 78). The primary substrates for STS are dihydroepiandrosterone sulphate and cholesterol sulphate, and additional substrates include pregnenolone sulphate and androstenediol-3-sulphate [[79]. The enzyme is widely distributed in human and fetal tissues including the brain, liver, adrenal cortex, placenta, skin, testes, ovaries and leucocytes [78]. The activity of STS in normal females is about 1.6 times that of normal males, due to only partial X inactivation of the STS gene locus [80,81].
In normal epidermis, the highest levels of STS activity are found in the granular and cornified cell layers [82]. Steroid sulphatase utilizes the lamellar body secretory system to translocate from the cytosol in the nucleated cell layers to the extracellular lamellar membrane domain in the stratum corneum [83]. Its substrate, cholesterol sulphate, also resides in the stratum corneum lamellar membranes and normally constitutes only 1–2% of the total lipid content. In the absence of STS, cholesterol sulphate accumulates, rising to 12% of the total lipid content, and the amount of cholesterol is correspondingly reduced [82]. This shift in lipid composition alters both the architecture and the physical properties of stratum corneum lamellar membranes, resulting in minor perturbations of epidermal barrier function [70].
Recessive X-linked ichthyosis is a classic example of retention hyperkeratosis; scaling results from impaired desquamation of corneocytes. At the ultrastructural level, this abnormal desquamation is explained by a failure of the normal, gradual and orderly degradation of corneodesmosomes, which instead persist into the outer stratum corneum [69,70]. Increased levels of cholesterol sulphate in the stratum corneum may be implicated in several different ways. First, degradation of corneodesmosomes requires the action of proteases that are inhibited by an acidic pH. Accumulation of cholesterol sulphate in RXLI lowers the stratum corneum pH [84]. Second, cholesterol sulphate has been shown to inhibit proteases [85], and reduced protease activity in RXLI stratum corneum has been documented [83]. Finally, cholesterol 3-sulphate interferes with cornified envelope assembly by diverting transglutaminase-1 activity from the formation of cross-links and esters to the hydrolysis of glutamine [86], but these results were obtained in an in vitro assay, and the cornified lipid envelope and underlying cornified cell envelope are ultrastructurally entirely normal [83].
Differential Diagnosis.
The differential diagnosis for RXLI includes ichthyosis vulgaris as well as mild forms of other generalized ichthyoses, such as lamellar ichthyosis. RXLI is usually distinguished from ichthyosis vulgaris by the presence of dark scale on the neck, prominent truncal involvement and scale of the lateral face, whereas hyperlinear palms, soles and lips, follicular keratosis and atopic eczema and asthma are more likely in ichthyosis vulgaris (see Table 121.3). The absence of a granular layer on histology is diagnostic for bialleic ichthyosis vulgaris, but when this finding is absent, histology does not rule in or out either diagnosis. In those cases with clinical and histological ambiguity, biochemical or genetic testing is indicated.
Diagnostic Options.
Presently, RXLI is often suspected prenatally due to low maternal uE3 levels obtained as part of a maternal ‘triple screen’ in the second trimester of pregnancy. Although cytogenetic testing is needed to rule out other more severe abnormalities, RXLI may be confirmed by several diagnostic means. Commonly employed tests are PCR or FISH [87]. These rapid and cheap DNA-based techniques take advantage of the high likelihood that RXLI is due to an STS gene deletion. However, they will not identify the minority of patients whose disease results from a point mutation in the STS gene, and sequencing or biochemical diagnostic techniques may be required in these instances.
Direct enzymatic assay of STS activity in cultured cells (e.g. leucocytes, fibroblasts, amniocytes) can be considered a gold standard since it will always identify enzymatic deficiency. It is widely available, reliable and sensitive to all genotypes [68,81]. One can also measure substrate accumulation in the blood by quantifying cholesterol sulphate levels [88]; unfortunately this clinical assay is not widely available. Historically, a qualitative estimate of high cholesterol sulphate levels via serum lipoprotein electrophoresis was used. Because the negatively charged cholesterol sulphate is predominantly carried in the low-density lipoprotein (LDL) fraction, it speeds the mobility of the LDL or β-lipoprotein fraction on serum lipoprotein electrophoresis [89].
Treatment.
Improving humidification of the skin may help those with mild RXLI, as evidenced by the propensity of the disease to improve in warm, humid climates. Urea, lactic acid, propylene glycol and retinoic acid containing emollients are highly effective. Because RXLI is a disease of abnormal desquamation, the use of α-hydroxy acids to help desquamate retained corneocytes is beneficial and usually well tolerated. Unfortunately, cholesterol-containing emollients are not commercially available. Topical retinoids can be helpful for localized, severe areas. Systemic retinoids may be beneficial, but the risk/benefit of using a systemic medication to treat a relatively mild skin condition must be considered.
Multiple Sulphatase Deficiency
Multiple sulphatase deficiency (MSD, OMIM 272200) is an extremely rare, autosomal recessive inborn error of metabolism, resulting from decreased activity of all sulphatase enzymes. The clinical phenotype of patients with this disorder combines the features of single sulphatase deficiencies: mucopolysaccharidosis (sulphatases for glycosaminoglycans), metachromatic leucodystrophy (sulphatases for glycolipids), RXLI (steroid sulphatase) and X-linked recessive chondrodysplasia punctata (arylsulphatase E) [90]. Features of MSD include abnormal facies (‘gargoylism’), hepatosplenomegaly, skeletal anomalies, mental retardation and neurological deterioration [91]. Although the rapidity of onset and severity of symptoms vary, infants who survive into childhood often develop an ichthyosis that is clinically similar to, but usually milder than, RXLI. The diagnosis of neonatal MSD should be considered when dealing with the association of distinct facial anomalies, limited joint mobility, ichthyosis and muscular hypotonia. Because MSD is an autosomal trait, both males and females are affected. The estimated frequency is 1 : 1.4 million.
Multiple sulphatase deficiency is caused by mutations in the SUMF1 gene on chromosome 3p26 [92,93] encoding the formylglycine-generating enzyme (FGE). Endoplasmic reticulum-localized FGE post-translationally activates sulphatases by oxidizing a specific cysteine residue generating formylglycine in their catalytic sites [94,95]. Individuals with SUMF1 mutations lack FGE and therefore have decreased activity of all sulphatases [96]. The accumulation of metabolic precursors in various tissues leads to the clinical manifestations of MSD.
References
1 Wells RS, Kerr CB. Genetic classification of ichthyosis. Arch Dermatol 1965;92:1–6.
2 Presland RB, Boggess D, Lewis SP, Hull C, Fleckman P, Sundberg JP. Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris. J Invest Dermatol 2000;115:1072–81.
3 Smith FJ, Irvine AD, Terron-Kwiatkowski A et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 2006;38:337–42.
4 Irvine AD, McLean WH. Breaking the (un)sound barrier: filaggrin is a major gene for atopic dermatitis. J Invest Dermatol 2006;126:1200–2.
5 Sandilands A, Terron-Kwiatkowski A, Hull PR et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet 2007;39:650–4.
6 Wells RS, Kerr CB. Clinical features of automal dominant and sex-linked ichthyosis in an English population. Brit Med J 1966;1:947–50.
7 Mevorah B, Krayenbuhl A, Bovey EH, Melle GD van. Autosomal dominant ichthyosis and X-linked ichthyosis. Comparison of their clinical and histological phenotypes. Acta Derm Venereol 1991;71:431–4.
8 Mevorah B, Marazzi A, Frenk E. The prevalence of accentuated palmoplantar markings and keratosis pilaris in atopic dermatitis, autosomal dominant ichthyosis and control dermatological patients. Br J Dermatol 1985;112:679–85.
9 Uehara M, Hayashi S. Hyperlinear palms: association with ichthyosis and atopic dermatitis. Arch Dermatol 1981;117:490–1.
10 Kuokkanen K. Ichthyosis vulgaris. A clinical and histopathological study of patients and their close relatives in the autosomal dominant and sex-linked forms of the disease. Acta Derm Venereol Suppl (Stockh) 1969;62:1–72.
11 Feinstein A, Ackerman AB, Ziprkowski L. Histology of autosomal dominant ichthyosis vulgaris and X-linked ichthyosis. Arch Dermatol 1970;101:524–7.
12 Fleckman P, Brumbaugh S. Absence of the granular layer and keratohyalin define a morphologically distinct subset of individuals with ichthyosis vulgaris. Exp Dermatol 2002;11:327–36.
13 Anton-Lamprecht I, Hofbauer M. [Ultrastructural distinctive features of autosomal dominant ichthyosis vulgaris and X-linked recessive ichthyosis]. Dermatologica 1972;145:60–4.
14 Sybert VP, Dale BA, Holbrook KA. Ichthyosis vulgaris: identification of a defect in synthesis of filaggrin correlated with an absence of keratohyaline granules. J Invest Dermatol 1985;84:191–4.
15 Fleckman P, Holbrook KA, Dale BA, Sybert VP. Keratinocytes cultured from subjects with ichthyosis vulgaris are phenotypically abnormal. J Invest Dermatol 1987;88:640–5.
16 Scott IR, Harding CR, Barrett JG. Histidine-rich protein of the keratohyalin granules. Source of the free amino acids, urocanic acid and pyrrolidone carboxylic acid in the stratum corneum. Biochim Biophys Acta 1982;719:110–17.
17 Scott IR, Harding CR. Filaggrin breakdown to water binding compounds during development of the rat stratum corneum is controlled by the water activity of the environment. Dev Biol 1986;115:84–92.
18 Scott IR. Alterations in the metabolism of filaggrin in the skin after chemical- and ultraviolet-induced erythema. J Invest Dermatol 1986;87:460–5.
19 Compton JG, DiGiovanna JJ, Johnston KA, Fleckman P, Bale SJ. Mapping of the associated phenotype of an absent granular layer in ichthyosis vulgaris to the epidermal differentiation complex on chromosome 1. Exp Dermatol 2002;11:518–26.
20 Hsu CK, Akiyama M, Nemoto-Hasebe I et al. Analysis of Taiwanese ichthyosis vulgaris families further demonstrates differences in FLG mutations between European and Asian populations. Br J Dermatol 2009;161:448–51.
21 Nomura T, Sandilands A, Akiyama M et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol 2007;119:434–40.
22 Chen H, Ho JC, Sandilands A et al. Unique and recurrent mutations in the filaggrin gene in Singaporean Chinese patients with ichthyosis vulgaris. J Invest Dermatol 2008;128:1669–75.
23 Weidinger S, O’Sullivan M, Illig T et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J Allergy Clin Immunol 2008;121:1203–9;e1201.
24 Schmuth M, Gruber R, Elias PM, Williams ML. Ichthyosis update: towards a function-driven model of pathogenesis of the disorders of cornification and the role of corneocyte proteins in these disorders. Adv Dermatol 2007;23:231–56.
25 Weiss G, Confino Y, Shemer A, Trau H. Cutaneous manifestations in the cardiofaciocutaneous syndrome, a variant of the classical Noonan syndrome. Report of a case and review of the literature. J Eur Acad Dermatol Venereol 2004;18:324–7.
26 Roberts A, Allanson J, Jadico SK et al. The cardiofaciocutaneous syndrome. J Med Genet 2006;43:833–42.
27 Okulicz JF, Schwartz RA. Hereditary and acquired ichthyosis vulgaris. Int J Dermatol 2003;42:95–8.
28 Koppe G, Marinkovic-Ilsen A, Rijken Y, De Groot WP, Jobsis AC. X-linked icthyosis. A sulphatase deficiency. Arch Dis Child 1978;53:803–6.
29 Lykkesfeldt G, Nielsen MD, Lykkesfeldt AE. Placental steroid sulfatase deficiency: biochemical diagnosis and clinical review. Obstet Gynecol 1984;64:49–54.
30 Ingordo V, D ’Andria G, Gentile C, Decuzzi M, Mascia E, Naldi L. Frequency of X-linked ichthyosis in coastal southern Italy: a study on a representative sample of a young male population. Dermatology 2003;207:148–50.
31 Sakura N, Nishimura SI, Matsumoto T, Ohsaki M. Frequency of steroid sulfatase deficiency in Hiroshima. Acta Paediatr Jpn 1998;40:63–4.
32 France JT, Liggins GC. Placental sulfatase deficiency. J Clin Endocrinol Metab 1969;29:138–41.
33 Shapiro LJ, Weiss R, Buxman MM et al. Enzymatic basis of typical X-linked ichthyosis. Lancet 1978;ii:756–7.
34 Hoyer H, Lykkesfeldt G, Ibsen HH, Brandrup F. Ichthyosis of steroid sulphatase deficiency. Clinical study of 76 cases. Dermatologica 1986;172:184–90.
35 Okano M, Kitano Y, Yoshikawa K, Nakamura T, Matsuzawa Y, Yuasa T. X-linked ichthyosis and ichthyosis vulgaris: comparison of their clinical features based on biochemical analysis. Br J Dermatol 1988;119:777–83.
36 Bradshaw KD, Carr BR. Placental sulfatase deficiency: maternal and fetal expression of steroid sulfatase deficiency and X-linked ichthyosis. Obstet Gynecol Surv 1986;41:401–13.
37 David M, Israel N, Merksamer R et al. Very low maternal serum unconjugated estriol and prenatal diagnosis of steroid sulfatase deficiency. Fetal Diagn Ther 1995;10:76–9.
38 Schoen E, Norem C, O’Keefe J, Krieger R, Walton D, To TT. Maternal serum unconjugated estriol as a predictor for Smith–Lemli–Opitz syndrome and other fetal conditions. Obstet Gynecol 2003;102:167–72.
39 Costagliola C, Fabbrocini G, Illiano GM, Scibelli G, Delfino M. Ocular findings in X-linked ichthyosis: a survey on 38 cases. Ophthalmologica 1991;202:152–5.
40 Lykkesfeldt G. Placental steroid sulphatase deficiency. Arch Dis Child 1985;60:595.
41 Jay B, Blach RK, Wells RS. Ocular manifestations of ichthyosis. Br J Ophthalmol 1968;52:217–26.
42 De Unamuno P, Martin-Pascual A, Garcia-Perez A. X-linked ichthyosis. Br J Dermatol 1977;97:53–8.
43 Traupe H, Happle R. Mechanisms in the association of cryptorchidism and X-linked recessive ichthyosis. Dermatologica 1986;172:327–8.
44 Lykkesfeldt G, Hoyer H, Lykkesfeldt AE, Skakkebaek NE. Steroid sulphatase deficiency associated with testis cancer. Lancet 1983;ii:1456.
45 Garcia Perez A, Crespo M. X-linked ichthyosis associated with hypertrophic pyloric stenosis in three brothers. Clin Exp Dermatol 1981;6:159–61.
46 Stoll C, Grosshans E, Binder P, Roth MP. Hypertrophic pyloric stenosis associated with X-linked ichthyosis in two brothers. Clin Exp Dermatol 1983;8:61–4.
47 Matsukura H, Fuchizawa T, Ohtsuki A et al. End-stage renal failure in a child with X-linked ichthyosis. Pediatr Nephrol 2003;18:297–300.
48 Ozawa H, Osawa M, Nagai T, Sakura N. Steroid sulfatase deficiency with bilateral periventricular nodular heterotopia. Pediatr Neurol 2006;34:239–41.
49 Mallory SB, Kletzel M, Turley CP. X-linked ichthyosis with acute lymphoblastic leukemia. Arch Dermatol 1988;124:22–4.
50 Doherty MJ, Glass IA, Bennett CL et al. An Xp;Yq translocation causing a novel contiguous gene syndrome in brothers with generalized epilepsy, ichthyosis, and attention deficits. Epilepsia 2003;44:1529–35.
51 Van Esch H, Hollanders K, Badisco L et al. Deletion of VCX-A due to NAHR plays a major role in the occurrence of mental retardation in patients with X-linked ichthyosis. Hum Mol Genet 2005;14:1795–803.
52 Chocholska S, Rossier E, Barbi G, Kehrer-Sawatzki H. Molecular cytogenetic analysis of a familial interstitial deletion Xp22.2-22.3 with a highly variable phenotype in female carriers. Am J Med Genet A 2006;140:604–10.
53 Thomas NS, Sharp AJ, Browne CE, Skuse D, Hardie C, Dennis NR. Xp deletions associated with autism in three females. Hum Genet 1999;104:43–8.
54 Brookes KJ, Hawi Z, Kirley A, Barry E, Gill M, Kent L. Association of the steroid sulfatase (STS) gene with attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2008;147B:1531–5.
55 Lynch HT, Ozer F, McNutt CW, Johnson JE, Jampolsky NA. Secondary male hypogonadism and congenital ichthyosis: association of two rare genetic diseases. Am J Hum Genet 1960;12:440–7.
56 Perrin JC, Idemoto JY, Sotos JF, Maurer WF, Steinberg AG. X-linked syndrome of congenital ichthyosis, hypogonadism, mental retardation and anosmia. Birth Defects Orig Artic Ser 1976;12:267–74.
57 Abe K, Matsuda I, Matsuura N, Murayama T, Uzuki K, Okuno A. X-linked ichthyosis, bilateral cryptorchidism, hypogenitalism and mental retardation in two siblings. Clin Genet 1976;9:341–5.
58 Yen PH, Allen E, Marsh B et al. Cloning and expression of steroid sulfatase cDNA and the frequent occurrence of deletions in STS deficiency: implications for X-Y interchange. Cell 1987;49:443–54.
59 Bick D, Curry CJ, McGill JR, Schorderet DF, Bux RC, Moore CM. Male infant with ichthyosis, Kallmann syndrome, chondrodysplasia punctata, and an Xp chromosome deletion. Am J Med Genet 1989;33:100–7.
60 Paige DG, Emilion GG, Bouloux PM, Harper JI. A clinical and genetic study of X-linked recessive ichthyosis and contiguous gene defects. Br J Dermatol 1994;131:622–9.
61 Hardelin JP, Levilliers J, Young J et al. Xp22.3 deletions in isolated familial Kallmann’s syndrome. J Clin Endocrinol Metab 1993;76:827–31.
62 Quinton R, Schofield JK, Duke VM et al. X-linked ichthyosis with hypogonadism: not always Kallmann’s syndrome. Clin Exp Dermatol 1997;22:201–4.
63 Ogata T, Petit C, Rappold G, Matsuo N, Matsumoto T, Goodfellow P. Chromosomal localisation of a pseudoautosomal growth gene(s). J Med Genet 1992;29:624–8.
64 Cuevas-Covarrubias SA, Gonzalez-Huerta LM. Analysis of the VCX3A, VCX2 and VCX3B genes shows that VCX3A gene deletion is not sufficient to result in mental retardation in X-linked ichthyosis. Br J Dermatol 2008;158:483–6.
65 Tarpey PS, Smith R, Pleasance E et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 2009;41:535–43.
66. Jamain S, Quach H, Betancur C et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 2003;34:27–9.
67 Kent L, Emerton J, Bhadravathi V et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet 2008;45:519–24.
68 Meyer JC, Groh V, Giger V, Weiss H, Varbelow H, Schnyder UW. Rapid laboratory diagnostic of X-linked ichthyosis. Dermatologica 1982;164:249–57.
69 Mesquita-Guimaraes J. X–linked ichthyosis. Ultrastructural study of 4 cases. Dermatologica 1981;162:157–66.
70 Zettersten E, Man MQ, Sato J et al. Recessive x-linked ichthyosis: role of cholesterol-sulfate accumulation in the barrier abnormality. J Invest Dermatol 1998;111:784–90.
71 Mevorah B, Frenk E, Muller CR, Ropers HH. X-linked recessive ichthyosis in three sisters: evidence for homozygosity. Br J Dermatol 1981;105:711–17.
72 Thauvin-Robinet C, Lambert D, Vaillant G et al. X-linked recessive ichthyosis in a girl: strategy for identifying the causal mechanism. Br J Dermatol 2005;152:191–3.
73 Mohandas T, Shapiro LJ, Sparkes RS, Sparkes MC. Regional assignment of the steroid sulfatase-X-linked ichthyosis locus: implications for a noninactivated region on the short arm of human X chromosome. Proc Natl Acad Sci U S A 1979;76:5779–83.
74 Bonifas JM, Morley BJ, Oakey RE, Kan YW, Epstein EH Jr. Cloning of a cDNA for steroid sulfatase: frequent occurrence of gene deletions in patients with recessive X chromosome-linked ichthyosis. Proc Natl Acad Sci U S A 1987;84:9248–51.
75 Shapiro LJ, Yen P, Pomerantz D, Martin E, Rolewic L, Mohandas T. Molecular studies of deletions at the human steroid sulfatase locus. Proc Natl Acad Sci U S A 1989;86:8477–81.
76 Bonifas JM, Epstein EH Jr. Detection of carriers for X-linked ichthyosis by Southern blot analysis and identification of one family with a de novo mutation. J Invest Dermatol 1990;95:16–19.
77 Ballabio A, Carrozzo R, Parenti G et al. Molecular heterogeneity of steroid sulfatase deficiency: a multicenter study on 57 unrelated patients, at DNA and protein levels. Genomics 1989;4:36–40.
78 Reed MJ, Purohit A, Woo LW, Newman SP, Potter BV. Steroid sulfatase: molecular biology, regulation, and inhibition. Endocr Rev 2005;26:171–202.
79 Ghosh D. Three-dimensional structures of sulfatases. Methods Enzymol 2005;400:273–93.
80 Migeon BR, Shapiro LJ, Norum RA, Mohandas T, Axelman J, Dabora RL. Differential expression of steroid sulphatase locus on active and inactive human X chromosome. Nature 1982;299:838–40.
81 Meyer JC, Gilardi S, Sigg C, Bruckner-Tudermann L. Intermediate levels of aryl sulfatase C in human leukocytes of female carriers for X-linked recessive ichthyosis. Arch Dermatol Res 1986;278:491–3.
82 Elias PM, Williams ML, Maloney ME et al. Stratum corneum lipids in disorders of cornification. Steroid sulfatase and cholesterol sulfate in normal desquamation and the pathogenesis of recessive X-linked ichthyosis. J Clin Invest 1984;74:1414–21.
83 Elias PM, Crumrine D, Rassner U et al. Basis for abnormal desquamation and permeability barrier dysfunction in RXLI. J Invest Dermatol 2004;122:314–19.
84 Ohman H, Vahlquist A. The pH gradient over the stratum corneum differs in X-linked recessive and autosomal dominant ichthyosis: a clue to the molecular origin of the ‘acid skin mantle’? J Invest Dermatol 1998;111:674–7.
85 Sato J, Denda M, Nakanishi J, Nomura J, Koyama J. Cholesterol sulfate inhibits proteases that are involved in desquamation of stratum corneum. J Invest Dermatol 1998;111:189–93.
86 Nemes Z, Demeny M, Marekov LN, Fesus L, Steinert PM. Cholesterol 3-sulfate interferes with cornified envelope assembly by diverting transglutaminase 1 activity from the formation of cross-links and esters to the hydrolysis of glutamine. J Biol Chem 2000;275:2636–46.
87 Valdes-Flores M, Kofman-Alfaro SH, Jimenez-Vaca AL, Cuevas-Covarrubias SA. Carrier identification by FISH analysis in isolated cases of X-linked ichthyosis. Am J Med Genet 2001;102:146–8.
88 Shackleton CH, Reid S. Diagnosis of recessive X-linked ichthyosis: quantitative HPLC/mass spectrometric analysis of plasma for cholesterol sulfate. Clin Chem 1989;35:1906–10.
89 Epstein EH Jr, Krauss RM, Shackleton CH. X-linked ichthyosis: increased blood cholesterol sulfate and electrophoretic mobility of low-density lipoprotein. Science 1981;214:659–60.
90 Castano Suarez E, Segurado Rodriguez A, Guerra Tapia A et al. Ichthyosis: the skin manifestation of multiple sulfatase deficiency. Pediatr Dermatol 1997;14:369–72.
91 Busche A, Hennermann JB, Burger F et al. Neonatal manifestation of multiple sulfatase deficiency. Eur J Pediatr 2008.
92 Cosma MP, Pepe S, Annunziata I et al. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 2003;113:445–56.
93 Dierks T, Schmidt B, Borissenko LV et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 2003;113:435–44.
94 Dierks T, Dickmanns A, Preusser-Kunze A et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 2005;121:541–52.
95 Dierks T, Schlotawa L, Frese MA, Radhakrishnan K, von Figura K, Schmidt B. Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann–Pick C1 disease – Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim Biophys Acta 2009;1793:710–25.
96 Schlotawa L, Steinfeld R, von Figura K, Dierks T, Gartner J. Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Hum Mutat 2008;29:205.
Keratodermic Ichthyoses
The term ‘keratodermic ichthyoses’ unfortunately has not entered the Ichthyosis Consensus Conference terminology. Nevertheless, the authors feel that this term is useful and helps clinicians in their diagnostic approach and with the classification of MEDOC. We classify this heterogeneous group as keratodermic ichthyoses based on their extensive keratotic scales and the presence of palmoplantar keratoderma. These disorders include keratinopathic ichthyoses, hystrix-type ichthyoses, KID (keratitis, ichthyosis and deafness) syndrome and erythrokeratoderma. Most of them are attributable to dominant-negative mutations in the genes encoding for keratins, connexins or loricrin. Thus, keratodermic ichthyoses include the previous group of autosomal dominant congenital ichthyoses. Here, only the keratinopathic ichthyoses are discussed. Please refer to Chapter 120 for other keratodermic ichthyoses.
Keratinopathic Ichthyoses
Keratinopathic ichthyoses are due to mutations in keratin genes (see Chapter 117). There are 54 functional human keratin genes; 21 of these, including epidermal and hair keratin genes, as well as hair follicle-specific epithelial keratins, have been associated with hereditary disorders [1]. When mutated, the normal tissue distribution of keratin expression has closely predicted the resultant disease phenotype. Germline mutations of the suprabasal epidermal keratins 1, 2 and 10 (KRT1, KRT2, KRT10) produce generalized MEDOC phenotypes characterized by the combination of hyperkeratosis and erosions or peeling (Fig. 121.3), whereas postzygotic mutations produce epidermolytic naevi that follow the lines of Blaschko (Fig. 121.4) [2]. Mutations in other epidermal keratins are associated with palmoplantar keratoderma and pachyonychia congenita, while abnormal basal layer keratins produce epidermolysis bullosa simplex. Although epidermolysis bullosa simplex is primarily a mechanobullous disorder, some forms also exhibit palmoplantar keratoderma. This section discusses the generalized epidermolytic disorders due to mutations in the outer epidermal keratins, whereas the mosaic forms, epidermolysis bullosa simplex, palmoplantar keratodermas, pachyonychia congenita and hair keratin disorders, are discussed elsewhere.
Fig. 121.3 Spectrum of the clinical phenotypes in epidermolytic ichthyosis. Upper and lower row shows patients with KRT10 and KRT1 mutations, respectively. Newborn infant a prone to widespread areas of denuded skin.
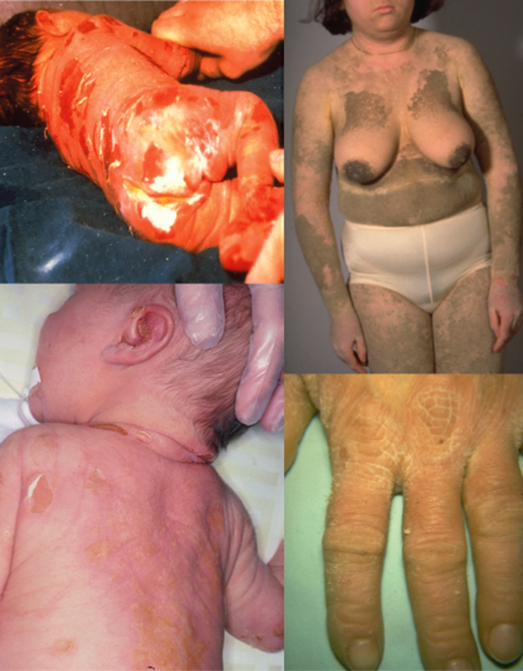
Fig. 121.4 Epidermolytic nevus due to post-zygotic mutations of KRT1 and KRT10 follows the lines of Blaschko and carries the risk of transmission of epidermolytic ichthyosis to offspring.
Epidermolytic Ichthyosis
Epidermolytic ichthyosis (EI; epidermolytic hyperheratosis, EHK; bullous congenital ichthyosiform erythroderma of Brocq, BCIE; OMIM 113800) is a severe congenital ichthyosis, characterized by erythroderma, blistering and hyperkeratosis. At birth it presents as enfant brûlé with widespread blisters. Following birth, blistering becomes less frequent and hyperkeratoses develop, typically with accentuation in the flexural areas. The disease was described first by Brocq in 1902 [3]. Subsequently, Lapière termed the disease ‘granular degeneration’, thereby describing his histological finding [4]. The term EHK was introduced by Frost and Van Scott about 10 years later [5], for the characteristic histopathological picture of massive hyperkeratosis overlying perinuclear vacuolization and lysis of the upper nucleated epidermal layers. This pattern is also observed in other keratin disorders and in some incidental settings, but when observed in the setting of a generalized ichthyotic phenotype, this histopathology is diagnostic. Epidermolytic ichthyosis is a rare, usually autosomal dominant skin disorder, estimated to occur in perhaps 1 in 100,000 to 300,000 births. Mutations in the KRT1 and KRT10 genes cause EI [6,7]. Depending on the mutations, varying degrees of clinical severity of blistering, keratoses and keratoderma can be seen in EI [8].
Clinical Features.
Epidermolytic ichthyosis usually presents at birth, with varying degrees of denuded skin, bullae, erosions and erythema (see Fig. 121.3). Neonates with EI resemble those with epidermolysis bullosa and other generalized neonatal blistering disorders, but close inspection may reveal areas of scaling or hyperkeratosis, providing a clue to the diagnosis. Blistering improves after the neonatal period, and subsequently hyperkeratosis rather than blistering becomes the predominant clinical feature. Children and adults, however, typically continue to exhibit signs of epidermal fragility, with blisters and erosions occurring at sites of minor trauma or with secondary infection.
The hyperkeratosis in EI is often most prominent overlying the joints and/or in the flexures. Typically, flexural surfaces have a ridged or verrucous pattern (see Fig. 121.3 and 121.4), whereas extensor surfaces have a characteristic cobblestone appearance. Although most patients have involvement of the entire body surface, some demonstrate sparing of the skin between the joints and on the trunk. The face is typically less involved than other sites, and ectropion does not occur. Scalp involvement is often present and may result in ‘nit-like’ encasement of the hair shaft.
Despite common clinical features, there is marked variable expression amongst individuals with EI and its variants [8]. The character of the hyperkeratosis may vary; some patients exhibit exuberant, spiky accumulations, whereas others have milder, flatter scale. The tendency to form a lichenified or ridged pattern, and for skin to be shed as full-thickness sheets of stratum corneum, however, is common to most phenotypes. This form of peeling has been termed Mauserung or ‘moulting effect’, and was originally described in association with ichthyosis bullosa of Siemens (superficial epidermolytic ichthyosis, SEI; see below) [9], but can be seen to some degree in other EI phenotypes. Some patients may have more marked fragility of the upper epidermis, with a tendency to develop deeper erosions with a moist, red base. Palmoplantar involvement ranges from complete sparing in some to severe, disabling PPK in others that is disproportionate to the severity of skin involvement elsewhere. Palmoplantar keratoderma can limit mobility of the digits and lead to joint contractures. Lordotic posture and gait abnormalities are likely due to limited joint extension. Likewise, some patients exhibit mild to moderate erythroderma, whereas others do not. DiGiovanna has observed that the nature of the mutations may predict the phenotype, since KRT1 mutations have been associated preferentially with PPK, whereas KRT10 mutations give rise to more non-palmoplantar variants [8]. As keratin 1 pairs with both keratin 9 and keratin 10 in palmoplantar skin, KRT1 mutations may be more deleterious in palmoplantar epidermis, whereas in the rest of the epidermis keratin 10 pairs with both keratin 1 and keratin 2, accounting for the more severe phenotype in these regions.
Another common characteristic of EI is a distinct malodour due to maceration of scales and bacterial overgrowth. This can be a significant problem for patients and their families, leading to social isolation. Other ectodermal structures such as hair, teeth and mucous membranes are unaffected. Nails are often normal as well, although a secondary dystrophy can develop due to chronic periungual inflammation or secondary dermatophyte infection. Other organ systems are not affected.
An annular variant of EI [10], which is also termed cyclic ichthyosis with EI, is rare and only seven families have been reported to date [1]. Affected individuals show redness and superficial erosions that resembles classic EI at birth. The clinical presentation improves during the first months of life, and later in life most individuals experience a benign course with localized disease. Flares of polycyclic psoriasiform plaques that persist for several weeks to several months are characteristic. Between the flares, the skin is normal except for hyperkeratoses on palms and soles. Mutations in the 2B domain of KRT10 have been discovered, suggesting that it represents a variant of EI [11], and this suggestion was further confirmed by the subsequent identification of KRT1 mutations [12].
Histopathological and Ultrastructural Features.
Epidermolytic ichthyosis is one of the few MEDOC in which the diagnosis can be confirmed by light microscopy. Histology reveals epidermolytic hyperkeratosis; i.e. acanthosis with perinuclear vacuolar changes, lysis and coarse basophilic clumps, resembling large keratohyalin granules, in the Malphigian layers, and marked hyperkeratosis. The degree of cytolysis ranges from subtle to extensive enough to result in intraepidermal blister formation with cleavage in the mid to upper epidermis. The histopathological findings of epidermolytic hyperkeratosis may be seen in some epidermal naevi and PPK, as well as in benign neoplasms such as epidermolytic acanthomas, or even as an incidental finding [13]. Clearly, a diagnosis of EI as a generalized MEDOC cannot be made without clinical and histopathological correlation.
Early electron microscopy by Weibel and Schnyder showed clumping of keratin filaments in the suprabasal layers of the epidermis leading to the hypothesis that a defect in tonofilament synthesis would lead to EHK [14]. These results were further substantiated 25 years later by the proof that these clumps contain keratin 1 and keratin 10 [15] and that KRT1 and KRT10 mutations cause EI.
Genetics and Pathogenesis.
Epidermolytic ichthyosis is due to mutations in the keratin 1 and keratin 10 proteins that are expressed suprabasally in the epidermis (genes localized to chromosomes 12q11-13 and 17q12-21, respectively) [6,16]. These mutations are primarily dominant, but rare recessive KRT10 mutations occur.
Keratins share a common domain structure with other intermediate filament proteins (see Chapter 117). They contain an alpha-helical central rod domain of ∼310 amino acids that is interrupted by non-helical linkers and flanked by amino-terminal head and carboxy-terminal tail domains. The central alpha-helical rod domain is composed of subdomains 1A, 1B, 2A and 2B, which are connected by three linkers, L1, L12 and L2. The rod domain is composed of seven-residue amino acid sequence repeats (a-b-c-d-e-f-g)n termed ‘heptad repeats’ with ‘a’ and ‘d’ being hydrophobic residues crucial for the formation of the coiled-coils (for recent reviews see references 1 & 17). The start of the 1A rod domain and the end of the 2B rod domain, the so-called helix initiation (HIP) and helix termination peptides (HTP), respectively, comprise short (∼20 amino-acid) sequence motifs that are highly conserved among the different keratins. These motifs play a pivotal role in overlap interactions in keratin intermediate filament (KIF) assembly, and thus mutations in these motifs interfere with the early stages of filament elongation. Keratins are obligate heterodimers, i.e. they constitute their filamentous stage by heteropolymeric pair formation of a type I and a type II keratin molecule. Compatible acidic type I and basic type II keratins combine to form heterodimers, and keratin 1 and keratin 10 form one such pair. These heterodimers then pair to form tetramers that further aggregate to form KIF.
The majority of mutations reported in keratins are missense mutations (∼90%) with a small number of small in-frame insertion/deletion mutations and a few intronic splice site defects leading to larger in-frame deletions. The functional consequence at the protein level is expression of mutant polypeptides with amino acid substitutions, deletions or insertion of a foreign amino acid at normal or near-normal levels. The mutant proteins form heterodimers with the wild-type keratin partner and thereby integrate into the keratin network where they render the cytoskeleton susceptible to collapse when exposed to environmental stress [18].
The cytoskeletal defect leads to a failure of lamellar body secretion with entombed lipid within corneocytes and a paucity of lamellar membranes in the intercellular domain of the stratum corneum [19]. This, in turn, results in a defective permeability barrier. Thus, despite the cellular fragility, lanthanum perfusion studies have demonstrated that the abnormal water permeation transits through the intercellular (and not via an intracellular) pathway. The basis for the ‘phenotypic shift’ from the neonatal blistering to the mature, hyperkeratotic phenotype probably, at least in part, resides in an increased demand for a competent barrier in a xerotic (postnatal) vs hydrated (intrauterine) environment, which in turn would stimulate homeostatic responses such as epidermal hyperplasia [20]. The amelioration of epidermal fragility postnatally may be a consequence of the upregulation of so-called wound-healing keratins (e.g. keratin 6 and keratin 16) [15,21], which do not bear mutations. The pathogenesis of the shift towards hyperkeratosis is probably more complex. Recent experiments indicate that mechanical stretching of keratinocytes expressing mutant KRT10 leads to p38 stimulation and diminished jun-kinase activation. Further, stressed KRT10 mutant keratinocytes release greater amounts of TNFα and RANTES whereas IL-1α secretion is reduced. This indicates that destabilization of the keratin intermediate filament network by mutant KRT10 affects stress signalling and innate immune responses of keratinocytes exposed to mechanical stretch [22].
Epidermolytic ichthyosis and related keratin disorders are mostly dominant-negative disorders but rare cases of recessive transmission occur. In the heterozygous state, the expression of the mutant gene product interferes with the function of the normal gene product. In general, disease-causing keratin mutations cluster at the HIP and HTP [23,24], but may also occur in the central linker region of the molecule or elsewhere. In keratinopathies, dominant-negative KRT mutations disrupt KIF assembly, resulting in clumped KIF, cytolysis and blister formation. More severe phenotypes of EI correlate with mutations in the HIP or HTP [24], but milder forms of EI may also have mutations in these regions [12,25], making exact genotype–phenotype correlations difficult.
The normal pattern of keratin expression correlates well with disease phenotypes of keratin disorders. KRT1 and KRT10 are solely expressed in suprabasal epidermis, thus mutations in both of them should present as identical phenotypes. However, as indicated above, in palmoplantar epidermis KRT9 pairs only with KRT1, and in non-palmoplantar epidermis KRT2 pairs only with keratin 10. Thus, mutations in the KRT1 gene tend to produce severe PPK accompanying moderate EI, whereas mutations in the KRT10 gene result in severe EI that rather spares the palms and soles (see Fig. 121.3) [8,26].
Differential Diagnosis and Diagnostic Considerations.
The widespread blistering and erosions seen in neonates with EI must be distinguished from blistering due to several other causes. Epidermolyis bullosa presents with widespread congenital erosions and is difficult to distinguish from neonatal EI. In epidermolyis bullosa, the non-eroded skin appears normal, whereas in EI, areas of hyperkeratosis may be seen. Skin biopsy to distinguish the two diseases may be necessary. Infections such as herpes simplex and varicella zoster viruses and staphylococcal scalded skin syndrome must be excluded by Giemsa-stained swab smears, culture and/or skin biopsy. In older children and adults with EI, the combination of hyperkeratosis with focal blistering is distinctive, and confirmation by the characteristic histopathology is readily available.
Superficial epidermolytic ichthyosis (SEI; see below) appears similar, although the erosions are generally more superficial and the hyperkeratosis is less marked and has a characteristic lichenified dark appearance notably on the neck and around flexures of the limbs. The distinction of mild EI from more severe cases of SEI on purely clinical grounds, however, may be difficult and molecular analysis is necessary for distinction [1].
Ichthyosis hystrix of Curth–Macklin (IHCM, see below) is a related keratinopathy, characterized by massive, warty hyperkeratosis on soles, palms, ankles, knees and elbows but lack of blistering. KRT1 mutations of the globular tail domain have been identified, suggesting that EI and IHCM are members of a genetic spectrum of phenotypes. This further strengthens the concept that patients with EI and PPK should be first screened for KRT1 gene mutations, whereas in those without PPK, the KRT10 gene should be examined first.
Prenatal Diagnosis.
Prenatal diagnosis can be obtained by genetic testing on cells collected in early gestation by chorionic villus sampling or amniocentesis [27]. Kindreds harbouring known mutations are the best candidates as screening may be directed towards a specific region of the gene. The ultrastructural feature of clumped KIF may be seen in amniotic fluid cells [28], providing an alternative means of prenatal diagnosis in families without documented mutations, albeit later in gestation than using DNA-based techniques. Historically, fetal skin biopsy was used successfully. However, the onset of cornification at 19 to 20 weeks is uncomfortably close to viability ages currently achievable [29].
Management.
Widespread blistering and denuded skin are therapeutic challenges specific to the newborn period in EI. Affected neonates require management in an intensive care nursery. Special care must be taken to prevent excessive fluid loss, which may lead to dehydration and electrolyte imbalances. Exposure to a high humidity environment and frequent use of emollients are helpful in this regard. Cutaneous fluid losses result in heat loss as well, and neonates with EI may have temperature instability. Erosions place these neonates at increased risk for skin and systemic infection, but because of excessive heat loss, signs of septicaemia may be masked. Bacterial swabs should be taken routinely, and prophylactic antibiotics considered until infection can be confidently ruled out. These neonates should be handled gently to minimize further skin injuries, using sterile techniques when possible, if widespread erosions are present. If avoidable, adhesives should not be applied. An impaired permeability barrier places these infants at increased risk for percutaneous absorption of potentially toxic substances, hence topical therapy should be limited to bland petrolatum-based emollients. Care must be taken to avoid contamination of emollient sources with repeated applications.
Blistering after the neonatal period may be indicative of secondary infection. Staphylococcus aureus is the most common organism, but in moist areas, Gram-negative bacteria may be causative. Culture and treatment with appropriate antibiotics are indicated. Superinfection with dermatophytes should be considered in patients with increased scale or pruritus and confirmed by KOH examination or culture. Topical antiseptics and antibacterial soaps may help to reduce secondary infections and control body odour from excessive bacterial colonization. Chronic suppressive antibiotic therapy is not recommended due to the potential for developing resistant pathogens.
The hyperkeratosis in EI presents a therapeutic challenge. Different types of humectants, emollients and keratolytics are used. However, individualized care is essential, because exfoliative products such as the α-hydroxy acids, lactic acid, propylene glycol and urea may be poorly tolerated due to burning or irritation through fissures and denuded areas. Generalized applications of salicylic acid should be avoided due to increased percutaneous absorption with risk of systemic toxicity. Topical retinoids and calcipotriol are also effective, but their use may be limited due to burning or irritation, as well as concern about toxicity from systemic absorption.
Oral retinoids (e.g. acitretin, previously etretinate) control hyperkeratosis very efficiently and are of high value in the treatment of this disfiguring disorder (Fig. 121.5). However, they may induce epidermal fragility and blister formation, particularly if used at higher doses. Moreover, due to the fragility of the upper Malphigian cell layers, the cleavage plane tends to occur at the stratum granulosum – stratum corneum interface, producing a tender base once the scale is shed. When employed, treatment should be initiated at a low dose (0.1 mg/kg) and increased gradually as tolerated (up to 0.5 mg/kg). A limited study of retinoid therapy for EI suggests that the treatment may be more effective for those with KRT10 gene mutations [21].
Fig. 121.5 A patient with epidermolytic ichthyosis caused by a severe KRT10 mutation before (left) and during treatment with etretinate.
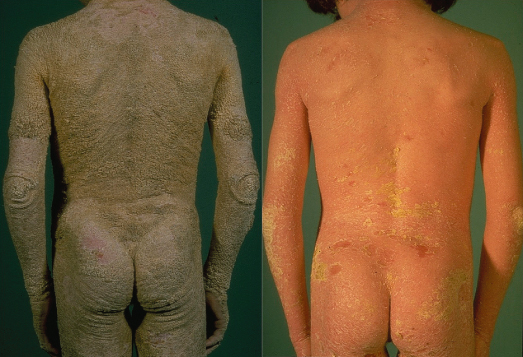
Superficial Epidermolytic Ichthyosis
Superficial epidermolytic ichthyosis (SEI; ichthyosis bullosa of Siemens, IBS; OMIM 146800) was originally described in 1937 as a unique, autosomal dominant form of bullous ichthyosis, distinct from that described by Brocq, affecting six members of a Dutch family. It received little attention until in 1986 Traupe and colleagues [9] characterized a second kindred affected with localized, superficial blistering, hyperkeratosis and lichenification. Superficial epidermolytic ichthyosis presents in infancy or early childhood with widespread blistering in response to trauma, heat or excessive sweating. Blistering improves with increasing age, becoming more localized to the acral extremities, and may be as mild as subtle areas of superficial erosions with collarettes of scale. Superficial peeling of the skin, described as Mauserung or ‘moulting effect’, is characteristic. Underlying redness is not prominent, although an erythrodermic infant with pustular lesions has been described [30]. Grey or brown hyperkeratosis and lichenification (Fig. 121.6) are most prominent at the elbows, wrists, knees, ankles, periumbilical region, buttocks and palms and soles.
Fig. 121.6 A patient with superficial epidermolytic ichthyosis caused by a KRT2 mutation. Note the superficial aspect of exfoliation known as ‘Mauserung’.
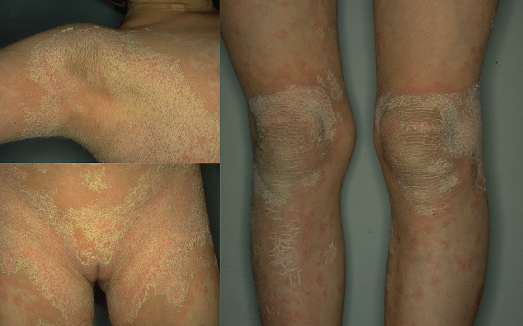
The histopathology of SEI shows epidermolytic hyperkeratosis confined to the uppermost stratum spinosum and granulosum with superficial blisters or polynuclear pustules. Electron microscopic analysis demonstrates slightly thickened bundles and discrete clumps of KIF in the upper Malpighian layers. This location is consistent with the tissue expression of KRT2. Mutations in the 1A and 2B domains of keratin 2 have been identified, but the majority of mutations affect the HTM of KRT2. One mutation, p.Glu487Lys, a potential methyl-CpG deamination, accounts for most cases and probably represents a mutational ‘hotspot’ [1].
The differential diagnosis of SEI includes disorders caused by suprabasal and superficial blistering such as EI, peeling skin syndrome and Comel–Netherton syndrome [31,32]. Severe forms of SEI are clinically similar to moderate EI, and in some cases, KRT2 mutations have been identified only after sequencing of the KRT1 and KRT10 genes did not reveal mutations [33,34]. Histological, ultrastructural and genetic analyses have shown that autosomal dominant ichthyosis exfoliativa is identical to SEI [35,36] and that this term should be discarded.
Because SEI is often mild and localized, treatment may not be necessary. Topical therapies, including the α-hydroxy acids, have a low risk of side-effects and may help control symptoms [37]. Low-dose oral retinoids are also effective in controlling vesiculation and scale [38], but their risks must be weighed against the potential benefits of therapy.
Peeling Skin Syndromes
Peeling skin syndromes (PSS) include a rare heterogeneous group of autosomal recessive genodermatoses characterized by an asymptomatic, superficial exfoliation of the outer epidermis, owing to a separation of the stratum corneum from the stratum granulosum at the subcorneal level [39,40]. Several types of PSS have been described [41–44], including generalized and acral forms. There are also cases in which acral and generalized skin peeling occur together [44]. Ultrastructural and light microscopic studies in both the generalized and the acral forms of the disorder show the same level of blistering in the epidermis, at the stratum granulosum–stratum corneum junction.
The acral peeling skin syndrome is a defined subtype (APSS; OMIM 609796) in which the peeling of the skin is limited to the dorsa of the hands and may be provoked by mechanical trauma or humidity [43,45]. This autosomal recessive disorder is due to TGM5 mutations [46]. Transglutaminase 5 (TG5) is strongly expressed in the epidermal granular cells, where it cross-links a variety of structural proteins in the terminal differentiation of the epidermis to form the cornified cell envelope. The exact pathomechanistic explanation of how TGM5 mutations lead to the phenotype remains to be identified, and we ignore which missing crosslinks are ultimately key for the development of the phenotype due to defective TG5. Some cases show cleavage of corneodesmosomes at the ultrastructural level [40], suggesting that hypothetically TG5 may be important for cross-linkage of corneodesmosomal proteins.
However, recent work reveals that such cases of peeling skin syndrome type B are also caused by mutations of cornedesmosin (CDSN) [47,48]. This autosomal-recessive inflammatory form of PSS (OMIM 270300) shows pruritic ichthyosiform erythroderma with generalized life-long patchy peeling and is sometimes associated with atopy and high IgE levels. Therefore, type B PSS has to be differentiated from Netherton syndrome with which it shares clinical features.
Ichthyosis Hystrix of Curth–Macklin
Ichthyosis hystrix is a descriptive term for a heterogeneous group of diseases characterized by severe, spiky and verrucous hyperkeratosis as seen in EI or erythrokeratodermia variabilis (EKV). In 1954, Curth and Macklin [49] described a family affected to varying degrees with PPK and symmetric involvement of dark, verrucous hyperkeratosis, a disorder now called ichthyosis hystrix of Curth–Macklin (IHCM; OMIM 146590). This rare, autosomal dominant disorder resembles EI, but lacks epidermal fragility. Patients are affected with PPK that is often mutilating and can have a striate pattern on the palms [50]. The degree of involvement elsewhere varies but tends to be more pronounced distally and, when severe, demonstrates exaggerated amounts of scale forming dark, pointy excrescences.
Routine histology shows papillomatous hyperplasia with hyperkeratosis and hypergranulosis. Electron microscopic findings are distinctive; KIF form continuous perinuclear shells, and perinuclear vacuoles and binucleate keratinocytes are seen [51]. In one family, a mutation in the variable tail region of the K1 gene has been identified, leading to cytoskeletal abnormalities that differ from those seen in EI, with impaired translocation of loricrin to the desmosomal plaques [50].
References
1 Arin MJ. The molecular basis of human keratin disorders. Hum Genet 2009;125:355–373.
2 Paller AS, Syder AJ, Chan YM, Yu QC, Hutton E, Tadini G, Fuchs E. Genetic and clinical mosaicism in a type of epidermal nevus. N Engl J Med 1994;331:1408–15.
3 Brocq L. Erythrodermie congenitale ichthyosiforme avec hyperepidermotrophie. Ann Dermatol Syph 1902;4:1–31.
4 Lapiere S. [A hereditary and familial series of Darier’s disease of bullous type; a case of ichthyosiform erythroderma of bullous type.] Arch Belg Dermatol Syphiligr 1953;9:249–53.
5 Frost P, Van Scott EJ. Ichthyosiform dermatoses. Classification based on anatomic and biometric observations. Arch Dermatol 1966;94:113–26.
6 Rothnagel JA, Dominey AM, Dempsey LD et al. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 1992;257:1128–30.
7 Cheng J, Syder AJ, Yu QC, Letai A, Paller AS, Fuchs E. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell 1992;70:811–19.
8 DiGiovanna JJ, Bale SJ. Clinical heterogeneity in epidermolytic hyperkeratosis. Arch Dermatol 1994;130:1026–35.
9 Traupe H, Kolde G, Hamm H, Happle R. Ichthyosis bullosa of Siemens: a unique type of epidermolytic hyperkeratosis. J Am Acad Dermatol 1986;14:1000–5.
10 Sahn EE, Weimer CE Jr, Garen PD. Annular epidermolytic ichthyosis: a unique phenotype. J Am Acad Dermatol 1992;27:348–55.
11 Joh GY, Traupe H, Metze D et al. A novel dinucleotide mutation in keratin 10 in the annular epidermolytic ichthyosis variant of bullous congenital ichthyosiform erythroderma. J Invest Dermatol 1997;108:357–61.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


