Diagnosis and Classification of Epidermolysis Bullosa
According to the most recent consensus on classification [3], there are four broad categories of EB, depending on the level of split within the skin (Fig. 118.2): EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and mixed EB (Kindler syndrome). Within each of these categories, there are subtypes that are clinically and sometimes genetically distinct. Molecular analyses of patients with different forms of EB over the last 15–20 years have demonstrated mutations in at least 13 different genes encoding protein components of the cutaneous basement membrane zone and desmosomes. Figure 118.3 shows the ultrastructural arrangement and important structural proteins at this site. Delineation of mutations in EB has had an impact on our understanding of this group of diseases in two main ways. First, as the number of mutations detected in these genes increases, it is becoming clearer that correlation can be made in many instances between genotype and phenotype. Second, knowledge of the molecular defect in each subtype of EB has enabled a more logical classification system to be drawn up than has been possible hitherto. Table 118.1 summarizes the classification of EB with the molecular defect in each form.
Table 118.1 Classification of major types of epidermolysis bullosa
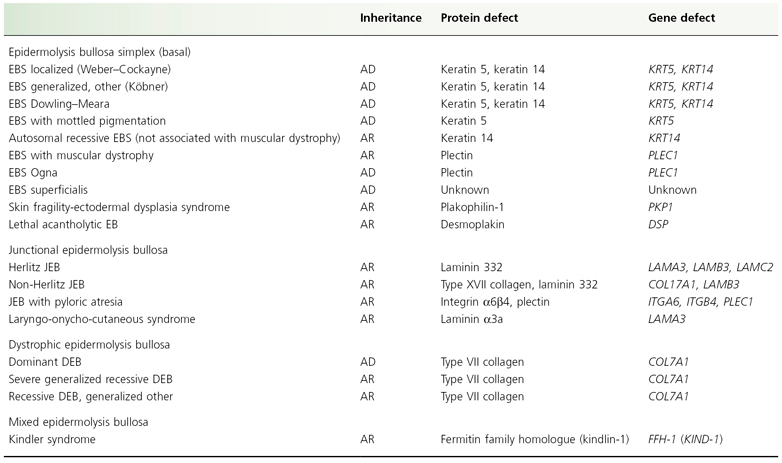
AD, autosomal dominant; AR, autosomal recessive.
Fig. 118.2 Ultrastructural planes of cleavage in different forms of EB. In EBS there is cytolysis of the basal keratinocytes, in JEB, blistering occurs at the level of the lamina lucida and in DEB, the split is beneath the lamina densa.
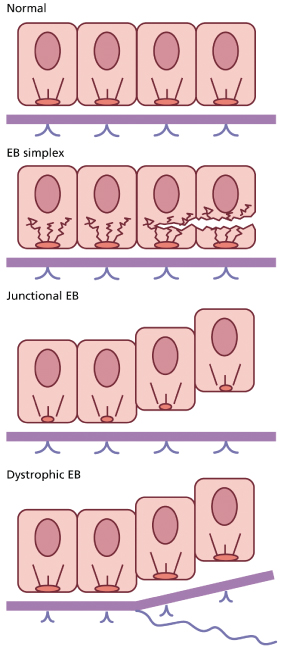
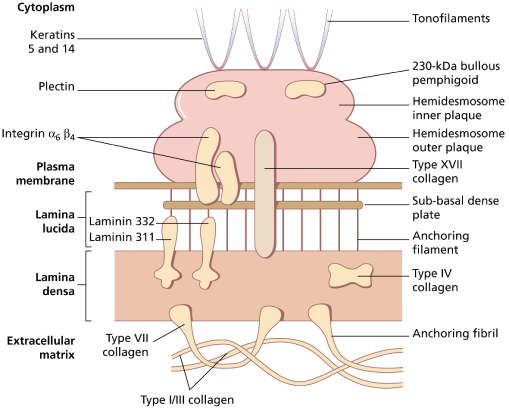
Obtaining an accurate diagnosis of the type of EB is exceptionally important to provide prognostic information, particularly in neonates in whom making an accurate diagnosis on the basis of clinical features alone may be impossible. The technique of taking skin biopsies in EB is of the greatest importance to provide useful tissue for the pathologist. Light microscopy will not delineate the precise plane of cleavage and therefore has no value in the diagnosis of EB. The biopsy should be taken from clinically unaffected skin. Disruption of the skin should be induced by rubbing the area to be biopsied with a finger or an India rubber for about 1 min. There should follow a delay of 5–10 min, after which the biopsy should be taken. In the authors’ view, a shave technique provides the best quality material because artefact is minimal, fixation is rapid, orientation is easier and healing is good. A useful technique is to pass a hypodermic needle in a shallow tangential direction through the biopsy site after local anaesthesia has been achieved, and then to cut along the upper surface of the needle with a scalpel blade. Punch biopsies are not ideal in EB since shearing stresses during sampling may mean that the epidermis becomes fully separated from the dermis, making subsequent interpretation difficult.
References
1 Priestley GC, Tidman MJ, Weiss JB et al. (eds). Epidermolysis Bullosa: A Comprehensive Review of Classification, Management and Laboratory Studies. Crowthorne: DEBRA, 1990.
2 Lin AN, Carter DM (eds). Epidermolysis Bullosa: Basic and Clinical Aspects. New York: Springer-Verlag, 1992.
3 Fine JD, Eady RAJ, Bauer EA et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008;58:931–50.
Epidermolysis Bullosa Simplex
Definition.
This is a group of inherited disorders characterized by mechanically induced blistering occurring within the epidermis itself. Although historically this has meant lysis of basal keratinocytes (basal EBS), the latest classification includes three disorders that result in a plane of separation within the suprabasal epidermis (suprabasal EBS). Because of the characteristic level of cleavage, EBS is sometimes termed epidermolytic EB.
There are several established variants, of which the following are the most important:
- EBS localized (previously Weber–Cockayne)
- EBS generalized, other (previously Köbner)
- EBS Dowling–Meara (herpetiformis).
In addition, there are a number of rarer variants that are encountered from time to time:
- EBS with muscular dystrophy [1]
- EBS with mottled pigmentation [2]
- autosomal recessive EBS (not associated with muscular dystrophy) [3,4]
- EBS Ogna [5]
- EB superficialis (suprabasal EBS) [6]
- Skin fragility-ectodermal dysplasia syndrome [7]
- Lethal acantholytic EB [8].
Aetiology and Pathogenesis.
The prevalence of the different forms of EBS has not been systematically studied and can therefore only be estimated; it probably varies from country to country [5,9–11]. The prevalence of localized EBS is estimated at about 5–20 per million. The ‘generalized, other’ form is rarer, possibly about 2 per million. With increasing experience of clinical and pathological diagnosis of the Dowling–Meara variant, the impression is that the disease is more common than previously believed, affected neonates being approximately as common as those with either dystrophic or junctional disease, with a prevalence probably in the region of 5–10 per million. The autosomal recessive types of EBS are probably extremely rare, but some of the rare forms may be locally common, for example the Ogna variant, whose prevalence may be as high as 14 per million in Norway [5].
Almost all forms of EBS are inherited as autosomal dominant traits, but some rare forms are inherited as autosomal recessive traits, notably EBS with muscular dystrophy [1,12] and autosomal recessive EBS caused by keratin 14 gene mutations [3,4].
Patients with the localized and generalized, other forms of EBS almost always have extensive family histories of the condition, and the occurrence of sporadic cases is relatively unusual. As is generally the case with dominantly inherited diseases, there may be considerable intrafamilial variation in severity. Approximately 50% of cases of Dowling–Meara EBS appear to be sporadic, caused by new autosomal dominant mutations.
Missense mutations in the genes encoding keratins 5 and 14 (KRT5 and KRT14, respectively), which constitute the predominantly expressed keratin pair in basal keratinocytes, have been identified in patients with the classic forms of EBS [13]. The position of mutations within these genes is important in determining the severity of the resulting disease: mutations in the highly conserved regions at the beginning and end of the central rod domain (the helix initiation and termination peptides, respectively) result in the more severe Dowling–Meara form of EBS, whereas mutations in less well-conserved regions of these genes cause localized or generalized, other EBS [14].
Pathology.
Epidermolysis bullosa is characterized at the pathological level by true epidermolysis, i.e. intracellular keratinocyte lysis. In basal forms of EBS, the ultrastructural level of lysis occurs in the basal keratinocytes [15] and in suprabasal forms, clefting is suprabasal or subcorneal (see below). Localized and generalized, other EBS cannot currently be distinguished ultrastructurally. However, Dowling–Meara EBS shows the distinctive feature of tonofilament clumping occurring within basal keratinocytes prior to their lysis [15], although this change cannot always be found easily, particularly in patients with clinical disease that is relatively mild. Prenatal diagnosis of EBS is discussed in Chapter 139.
Clinical Features
Localized (Weber–Cockayne) Epidermolysis Bullosa Simplex
This variant of EBS most characteristically has its onset in early childhood, but very often not until walking is established. In some cases, the condition is not revealed until adolescence or early adult life, when the subject is required to undertake unaccustomed activity, for example a forced march in the army.
Those who have localized EBS often do not consider themselves to have a medical problem, merely an exaggeration of the normal tendency to blister during or after hard walking or running, or after intensive use of the hands. Rubbing of the feet by footwear is generally the major cause of blisters, and the most common parts of the feet to be affected are the soles and the junctions between the sole and the sides of the toes or the main part of the foot (Fig. 118.4). A particular feature of all types of EBS is a progressively increased tendency to blister as the environmental temperature rises. So great is this effect that some patients who have marked disability in hot summer weather may have little or none in the winter. Although it is sometimes implied that EBS of this type is a relatively trivial disease, some patients experience very substantial disability, mainly because of difficulty in walking. Some individuals may be able to walk only 100 or 200 m on a summer day before painful blistering occurs. Other patients may have problems with manual tasks and find it impossible to use hand tools for more than brief periods.
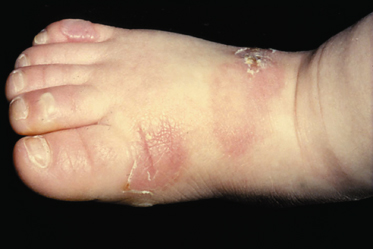
Blisters tend to be small, up to about 2 cm in diameter, and despite their relatively superficial localization they are generally tense (Fig. 118.5); they may even be haemorrhagic. A particular feature is an erythematous halo around the blisters, a characteristic that is generally lacking in other types of EB in the absence of secondary infection. Nevertheless, secondary infection is common once blisters have ruptured. Although the blisters will heal rapidly in the absence of secondary infection, the frequent recurrence of provocative trauma at affected sites tends to cause new blisters to occur underneath and at the margins of ones that are in the process of healing. A degree of hyperkeratosis often marks sites of recurrent blistering. The nails are not usually affected unless subject to considerable trauma, such as a heavy object being dropped on to toenails or toenails being trodden on. Areas of the body other than the hands and feet may be affected, for example, under elastic in clothing. The mouth is almost always spared.
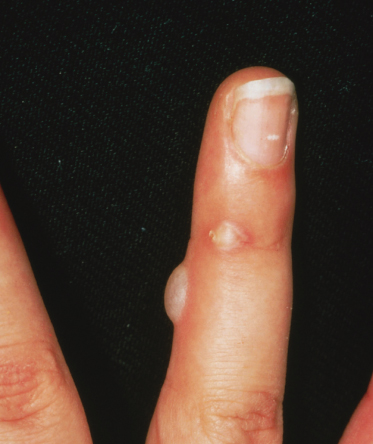
Generalized, Other (Köbner) Epidermolysis Bullosa Simplex
Generalized, other EBS tends to have an early onset, either during the perinatal period or during the first few months of life. It is not infrequently already present at birth.
In the perinatal period, blistering and erosions occur at sites determined by trauma during delivery and by handling in the nursery. Lesions heal quickly without scarring. Thereafter, the rate of new blister formation tends to slow down, with new lesions appearing only at sites of continuing friction, particularly in the napkin area. Oral lesions do occur, but tend not to be prominent and only rarely interfere with feeding. Nail involvement is unusual and when nails are occasionally shed following subungual blistering, they generally regrow without dystrophy.
Rubbing of the skin tends to be the main provocative factor. The blistering tendency is much more apparent in warm weather, to the extent that some patients’ problems may be more or less confined to the summer months. As the child starts to crawl, lesions may occur on the knees, feet, elbows and hands. With the onset of walking, the principal problem localizes to the feet and ankles, with the hands being the next most frequently affected site. In adults, it is rare for lesions to occur elsewhere, although blisters can arise at any site under appropriate provocation.
The blisters are tense and occur at the same sites on the hands and feet as in localized EBS. Likewise, secondary infection is perhaps the principal complication of the disorder. In the absence of such infection, the blisters heal fairly rapidly without scarring.
Dowling–Meara Epidermolysis Bullosa Simplex [16–19]
Clinically, Dowling-Meara EBS generally causes blistering with an onset in early infancy. There is a great range of severity in individual cases. Blistering may be exceptionally severe during the neonatal period to the extent that death may occur, usually as a result of sepsis. In the severe case, blistering may appear to arise quite spontaneously, particularly in a hot environment. Blisters may be even more often haemorrhagic than in other forms of EBS, and milia may be a transient feature after blisters have healed. It is important to be aware that milia are not pathognomonic of dystrophic forms of EB, and that they may occur, albeit rather fleetingly, in all the other forms, but perhaps particularly in Dowling–Meara EBS.
The hands and feet are the sites of predilection (Fig. 118.6), even in the neonatal period, and blisters at these sites are similar to those seen in other forms of EBS. However, it is particularly characteristic for blistering on the palms and soles to be succeeded by focal keratoderma, although this is also seen, usually to a lesser degree, in other types of EBS. On occasions, this keratoderma may be very prominent and associated with flexion deformity and loss of function. Children with Dowling–Meara EBS will often have considerable discomfort of their feet and the onset of walking is frequently delayed as a result.
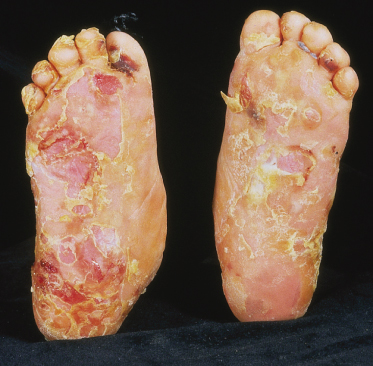
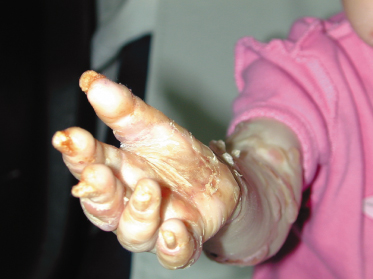
A rather characteristic thickening of the nails is also commonly seen in Dowling–Meara EBS (Fig. 118.7). This may be present in the neonatal period and in combination with thickening of palmar and plantar skin, this can be diagnostically helpful.
Fig. 118.7 Dowling–Meara EBS, showing palmar hyperkeratosis, nail dystrophy and blisters on the arm.
Blisters frequently occur at other sites on the face (Fig. 118.8), trunk and limbs, and tend to be disposed in groups with an erythematous border, hence the original name, EB ‘herpetiformis’. However, these groups are perhaps more often annular or arcuate than truly herpetiform. Of these other sites, the neck and axillae are particularly commonly affected. A major provocative factor appears to be friction from the seams of clothing. However, in this condition, groups of blisters may appear with remarkably little provocation. High environmental temperatures seem to be of great importance in reducing the threshold for blistering. Like other types of EBS, secondary infection is very common.
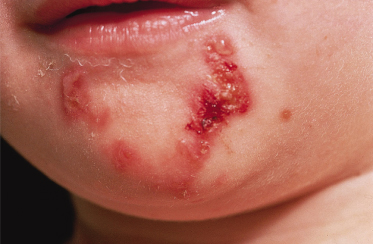
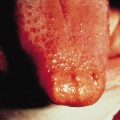
Oral involvement is usually not prominent. However, a proportion of severely affected neonates may experience extreme oropharyngeal blistering with potentially serious interference with feeding. These babies may have inco-ordination of swallowing, with a tendency to aspirate feeds, and they frequently also demonstrate marked gastro-oesophageal reflux.
Hoarseness of the voice is quite often present, particularly in the more severely affected case; a weak cry may be noticeable in the neonatal period. Laryngeal involvement was previously considered virtually pathognomonic of the Herlitz type of JEB, but it is now clear that it also occurs regularly in Dowling–Meara EBS.
Prognosis.
Generally, the prognosis in EBS is good, particularly in the common localized type, with the great majority of patients having a normal life expectancy. However, disability can be significant, with patients’ choices of career, housing, employment and leisure activity being constrained by limitations on the distance they can walk. Although the Dowling–Meara type of EBS can undoubtedly be lethal in early infancy, the blistering tendency will improve with time, and some adults who had problems with the condition as children later become more or less free from any evidence of the disease. However, other adults remain substantially disabled by Dowling–Meara EBS throughout their lives [20], particularly as a result of recurrent blistering of the hands and feet, and palmar and plantar keratoderma.
Differential Diagnosis.
The principal problem in diagnosis of the more common varieties of EBS is to distinguish them clinically from other forms of EB, although, in the main, this is a problem only in the neonatal period. Immunohistochemistry and electron microscopy of appropriate skin biopsies usually allow differentiation, but experience is required for reliable interpretation.
In the neonate, EB may be confused with any of the following: sucking blisters, incontinentia pigmenti, miliaria crystallina, bullous ichthyosiform erythroderma, bullous impetigo, staphylococcal scalded skin syndrome, EEC (ectodactyly–ectodermal dysplasia and clefting) syndrome, AEC (ankyloblepharon ectodermal defects and clefting) syndrome, neonatal or congenital varicella and herpes simplex, neonatal pemphigus or pemphigoid gestationis, infantile pemphigoid, bullous mastocytosis, cutis aplasia (including focal dermal hypoplasia) and transient neonatal porphyrinaemia. In adults, pachyonychia congenita will need to be considered, particularly in Dowling–Meara EBS when palmar and plantar hyperkeratosis and nail thickening may both be prominent.
Treatment.
In localized and generalized, other EBS, a long family association with the disease, combined with an awareness of both the provoking influences and the limitations of available therapy, make it relatively unusual for patients to seek medical assistance. When they do, the most useful contribution one can make is to provide advice about suitable footwear, general care of the feet, pain management and genetic counselling.
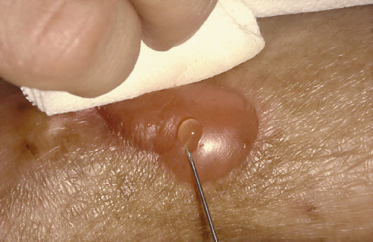
Fresh blisters should be drained after puncturing them with a sterile disposable needle, as they tend to extend if left alone (Fig. 118.9). When blisters tend to refill after simple puncturing, a small V-shaped incision with a pair of sharp sterile scissors may be more effective in ensuring decompression of the blister. If the patient finds decompression painful after lancing the blister, aspiration can be performed using a fine-gauge needle and syringe. The blister roof should be left in situ. It is often useful to bathe blistered feet and hands in warm water containing potassium permanganate at a dilution of about 1:8000. Currently available dressings for erosions and blisters in patients with EBS are not always ideal for meeting their requirements. Patients and their parents generally make up their own minds about the dressing materials that most suit their own needs, and the dermatologist’s priority is first to ensure that they are properly informed about the range of different types of dressing available and, second, that they are able to secure a supply of their chosen dressings as economically as possible.
Fig. 118.9 Blisters should be pierced with a sterile hypodermic needle, the contents expressed and the blister roof left in situ.
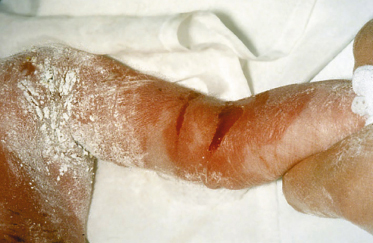
Patients often like to use topical antimicrobial applications because they are aware of the frequent occurrence of secondary bacterial infection. However, the regular use of any particular preparation tends to be associated with the development of bacterial resistance to the antimicrobial employed. The authors tend to recommend antimicrobial rather than antibiotic topical preparations. Lipid-stabilized 1% hydrogen peroxide cream (Crystacide®, GP Pharma) is generally well tolerated and can be used regularly without risk of resistance. The application of culinary-grade cornflour to eroded areas in EBS may be particularly helpful in drying the lesions and appears to hasten healing (Fig. 118.10); when blistering is predominantly on the feet, this may be applied directly into the socks before they are put on. Cornflour used in this way may also reduce the occurrence of new friction-induced blisters.
Fig. 118.10 Neonate with Dowling–Meara EBS. Cornflour is used to dry the blisters and reduce further friction.
Suitable dressings for those with EBS include hydrogels such as Actiform Cool® (Activa) and Intrasite Conformable® (Smith & Nephew) which have a cooling effect which offers comfort. Suprasorb X® (Activa) is a cellulose dressing which is very soft and conformable, and provides pain relief. Suprasorb X + PHMB® (Activa) has additional antimicrobial action for those prone to colonization or infection.
Foam dressings such as Mepilex®, Mepilex Lite® and Mepilex Transfer® (Mölnlycke Healthcare) are favoured by some, although these tend to increase heat so may not be well tolerated. Bordered dressings such as Mepilex Border Lite® (Mölnlycke Healthcare) and Alleyvn Gentle Border® (Smith & Nephew) are useful for isolated blister sites. However, many prefer not to use foam dressings as they find these exacerbate blistering.
It is important to provide children with EBS with footwear that allows them the maximum mobility while providing the best possible protection for their feet. Many children can wear ‘off-the-peg’ shoes if these incorporate appropriate design features. Ideally, these shoes are made of very soft leather, with the minimum number of internal seams. There should be plenty of room for the toes, in both the horizontal and vertical planes. Both the uppers and the insole should be made of permeable leather, in order to keep the foot as cool and dry as possible, and the inside of the sole should have a shape that is as anatomically appropriate as possible. However, few ‘off-the-peg’ shoes fulfil all these criteria. Socks should be absorbent and should therefore contain a high proportion of cotton. They should also provide additional cushioning. Towelling types of sports socks are ideal. It is sometimes useful for the patient to wear two pairs of socks as this helps to reduce friction. Socks containing a silver thread help to keep the feet cool and reduce the bacterial load. Suppliers of silver socks include Carnation Footcare and Best 4 Feet. DermaSilk Therapeutic Clothing supply socks which help to keep the feet cool and reduce friction.
Many babies and young children with generalized and Dowling–Meara EBS are very fragile, and will need protective measures and careful handling as described below under DEB.
It is very important to avoid the use of heavy dressings, which may increase the skin surface temperature and therefore the rate of blistering. In children with Dowling–Meara EBS, it seems especially important to check that clothing does not have rough internal seams and that it fits loosely, especially at the neck, wrists and ankles. Similarly, edges of dressings frequently lead to blistering: applying a layer of Aquacel hydrofiber® dressings (Convatec) between the skin and the edge of the chosen dressing should reduce this risk.
Avoidance of high environmental temperatures whenever possible is a helpful measure, and it is especially important to keep affected infants cool. The authors’ practice is to use an air-conditioned cubicle for these infants during any admission to hospital, and severely affected infants may benefit from a portable air-conditioning unit for use at home in the early years.
Topical application of 10% glutaraldehyde or 10% aluminium chloride hexahydrate has been advocated for the soles of patients with localized EBS [21,22] but, in practice, neither has proven particularly useful for the majority of patients. Many patients with EBS, especially the localized type, complain of hyperhidrosis of the feet, which exacerbates the blistering tendency, particularly in hot weather. These patients may derive some benefit from iontophoresis, using either plain tap water or water with the addition of the anticholinergic agent glycopyrronium bromide.
The recurrent painful blistering and plantar hyperkeratosis that arise in EBS, particularly in the Dowling–Meara form, often significantly limit mobility. The authors have found the use of low-dose amitriptyline (starting at 0.5 mg/kg per day and increasing according to response) to be particularly useful in reducing discomfort and thereby increasing distances that patients are able to walk.
The authors have occasionally used short courses of oral prednisolone to reduce blistering temporarily in Dowling–Meara EBS. The effectiveness of oral corticosteroid therapy in localized EBS was documented many years ago [23] and may be worth considering as a short-term measure when symptoms are particularly distressing in any form of EBS.
Some neonates severely affected with Dowling–Meara EBS have required nasogastric feeding for a period which may last several months. The authors recommend using a long-term feeding tube to reduce trauma. The tube can be secured using a silicone-based tape such as Mepitac® (Mölnlycke Healthcare) or Siltape® (Advancis Medical). If an adhesive tape is required it should be removed using a silicone medical adhesive remover such as Appeel® (Clinimed) or Niltac® (Trio Healthcare) in order to avoid skin stripping as the tape is removed.
Rarer Types of Epidermolysis Bullosa Simplex
Epidermolysis Bullosa Simplex with Muscular Dystrophy
This is a rare form of autosomal recessive EBS that results from mutations in the gene encoding plectin (PLEC1), a cytoskeleton membrane-anchoring protein [1,24,25]. Generalized blistering is of early onset, with an acral preponderance, atrophic scarring and nail dystrophy [26,27]. Laryngeal involvement may give rise to considerable respiratory compromise [28]. As with other forms of EBS, focal palmar and plantar hyperkeratosis tends to develop at sites of previous blistering. Progressive muscular dystrophy may very rarely be present at birth, but more commonly starts anywhere between the first year and the fourth decade of life, reflecting the important role of plectin in skeletal muscle as well as in skin.
Epidermolysis Bullosa Simplex with Mottled Pigmentation [29–31]
This autosomal dominant form of EBS is characterized by mechanically induced blistering, which resembles generalized, other EBS clinically, healing without scarring or atrophy. The mucosae are generally not affected. Blistering becomes less prominent with increasing age and may even disappear. Pigmentary abnormalities are the main distinguishing feature of the disorder, and take the form of well-demarcated pigmented macules 2–5 mm in diameter, most profuse on the trunk and the proximal limbs, which may be present from very early in life and whose appearance does not seem to be a direct result of blistering. In several cases, there may be a mixture of hyper- and hypopigmented macules. Punctate palmoplantar keratoderma and nail abnormalities are also common. Molecular analysis has demonstrated heterozygosity for a specific missense mutation in the keratin 5 gene in the vast majority of patients [30,31], although a keratin 14 mutation has also been identified in this condition [32].
Autosomal Recessive Epidermolysis Bullosa Simplex
There are rare families with autosomal recessive EBS in which there is generalized blistering that heals without scarring. In one severely affected pedigree from Sudan, there was also anaemia, involvement of the upper airway and death in early childhood [3]. Homozygosity for ‘knockout’ mutations of the keratin 14 gene, KRT14, has been identified in some families [33–36], and a homozygous missense mutation of keratin 14 in a family with a milder EBS localized-like phenotype was also found [37].
Epidermolysis Bullosa Simplex Ogna Type
This subtype of autosomal dominant EBS, characterized by blistering on the hands and feet predominantly and the presence of bruising, has been described in a large Norwegian pedigree [5]. Recently, the mutation in this family, and in a further German family with features of this form of EBS, has been identified as a specific mis-sense mutation in the rod domain of plectin [38]. Unlike recessive EBS caused by plectin mutations, neuromuscular involvement is not a feature of Ogna EBS.
Epidermolysis Bullosa Superficialis [6]
This is a rare autosomal dominant form of EBS with the plane of blistering just beneath the stratum corneum. It is not associated with scarring, but milia and nail dystrophy may be present. The molecular basis for EB superficialis is not currently known and it is not certain that it is a distinct entity.
Skin Fragility-Ectodermal Dysplasia Syndrome [7]
Skin fragility-ectodermal dysplasia syndrome is a rare form of suprabasal EBS characterized by sparse hair, nail dystrophy, dental abnormalities, skin fragility, focal palmoplantar keratoderma and, in some cases, reduced sweating [7,39]. It results from autosomal recessive mutations in the plakophilin 1 gene (PKP1), a desmosomal armadillo protein involved in maintaining structural integrity of desmosomes as well as having a role in signal transduction. Skin biopsies show suprabasal acantholysis and widening of intercellular spaces. Ultrastructurally, desmosomes have a ‘pinched off’ appearance and lack the normal association with keratin intermediate filaments. Plakophilin-1 immunostaining is markedly reduced or absent.
Lethal Acantholytic Epidermolysis Bullosa [8]
This autosomal recessive form of suprabasal EBS has been described in a single proband who had extreme skin fragility, nail loss, alopecia and natal teeth, with death occurring in the neonatal period. This arose from compound heterozygosity for mutations in the desmoplakin gene (DSP) which resulted in loss of the tail domain.
References
1 Smith FJD, Eady RAJ, Leigh IM et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nature Genet 1996;13:450–7.
2 Fischer T, Gedde–Dahl T. Epidermolysis bullosa simplex and mottled pigmentation: a new dominant syndrome. Clin Genet 1979;15:228–38.
3 Salih MAM, Lake BD, El Hag MA et al. Lethal epidermolytic epidermolysis bullosa: a new autosomal recessive type of epidermolysis bullosa. Br J Dermatol 1985;113:135–43.
4 Fine J-D, Johnson L, Wright T et al. Epidermolysis bullosa simplex: identification of a kindred with autosomal recessive transmission of the Weber–Cockayne type. Pediatr Dermatol 1989;6:1–5.
5 Gedde-Dahl T. Epidermolysis Bullosa. A Clinical, Genetic and Epidemiological Study. Baltimore: Johns Hopkins Press, 1971.
6 Fine J-D, Johnson L, Wright T. Epidermolysis bullosa simplex superficialis. A new variant of epidermolysis bullosa characterized by subcorneal skin cleavage mimicking peeling skin syndrome. Arch Dermatol 1989;125:633–8.
7 McGrath JA, McMillan JR, Shemanko CS et al. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet 1997;17:240–4.
8 Jonkman MF, Pasmooij AM, Pasmans SG et al. Loss of desmoplakin tail causes lethal acantholytic epidermolysis bullosa. Am J Hum Genet 2005;77:653–60.
9 Horn HM, Priestley GC, Tidman MJ. Epidemiology of epidermolysis bullosa in Scotland. Br J Dermatol 1995;133:1005.
10 Kero M. Occurrence of EB in Finland. Acta Dermatol Venereol (Stockh) 1984;64:57–62.
11 Fine J-D, Bauer EA, McGuire J et al. (eds). Epidermolysis Bullosa. Clinical, Epidemiologic and Laboratory Advances and the Findings of the National Epidermolysis Bullosa Registry. Baltimore: Johns Hopkins University Press, 1999.
12 Fine J-D, Stenn J, Johnson L et al. Autosomal recessive epidermolysis bullosa simplex: generalized phenotypic features suggestive of junctional or dystrophic EB, and association with neuromuscular disease. Arch Dermatol 1989;125:931–8.
13 Lane EB. Keratin diseases. Curr Opp Genet Dev 1994;4:412–18.
14 Fuchs E. Genetic disorders of keratins and their associated proteins. J Dermatol Sci 1996;13:181–92.
15 Haneke E, Anton–Lamprecht I. Ultrastructure of blister formation in epidermolysis bullosa hereditaria. V. Epidermolysis bullosa simplex localisata type Weber–Cockayne. J Invest Dermatol 1982;164:219–23.
16 McGrath JA, Ishida-Yamamoto A, Tidman MJ et al. Epidermolysis bullosa simplex (Dowling–Meara): a clinicopathological review. Br J Dermatol 1992;126:421–30.
17 Buchbinder LH, Lucky AW, Ballard E et al. Severe infantile epidermolysis bullosa simplex Dowling–Meara type. Arch Dermatol 1986;122:190–8.
18 Hachem-Zadeh S, Rappersberger K, Livshin R et al. Epidermolysis bullosa herpetiformis Dowling–Meara in a large family. J Am Acad Dermatol 1988;18:702–6.
19 Furumura M, Imayama S, Hori Y. Three neonatal cases of epidermolysis bullosa herpetiformis (Dowling–Meara type) with severe erosive skin lesions. J Am Acad Dermatol 1993;28:859–61.
20 McGrath JA, Burrows NP, Russell-Jones R et al. Epidermolysis bullosa simplex Dowling–Meara: troublesome blistering in an adult patient. Dermatology 1993;186:68–71.
21 DesGroseilliers J-P, Brisson P. Localized epidermolysis bullosa: report of two cases and evaluation of therapy with glutaraldehyde. Arch Dermatol 1974;109:70–2.
22 Tkach JR. Treatment of recurrent bullous eruption of the hands and feet (Weber–Cockayne disease) with topical aluminium chloride. J Am Acad Dermatol 1982;6:1095–6.
23 Readett MD. Localized epidermolysis bullosa. BMJ 1961;1:1510–11.
24 McLean WHI, Pulkinnen L, Smith FJD et al. Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organisation. Genes Dev 1996;10:1724–35.
25 Gache Y, Chavanas S, Lacour JP et al. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest 1996;97:2299–307.
26 Fine J-D, Stenn J, Johnson L et al. Autosomal recessive epidermolysis bullosa simplex. Arch Dermatol 1989;125:931–8.
27 Niemi K-M, Sommer H, Kero M et al. Epidermolysis bullosa simplex associated with muscular dystrophy with recessive inheritance. Arch Dermatol 1988;124:551–4.
28 Mellerio JE, Smith FJD, McMillan JR et al. Recessive epidermolysis bullosa simplex associated with plectin mutations: infantile respiratory complications in two unrelated cases. Br J Dermatol 1997;137:898–906.
29 Bruckner-Tuderman L, Vogel A, Ruegger S et al. EB simplex with mottled pigmentation. J Am Acad Dermatol 1989;21:425–32.
30 Uttam J, Hutton E, Coulombe PA et al. The genetic basis of epidermolysis bullosa simplex with mottled pigmentation. Proc Natl Acad Sci USA 1996;93:9079–84.
31 Irvine AD, McKenna KE, Jenkinson H et al. A mutation in the V1 domain of keratin 5 causes epidermolysis bullosa simplex with mottled pigmentation. J Invest Dermatol 1997;108:809–10.
32 Horiguchi Y, Sawamura D, Mori R et al. Clinical heterogeneity of 1649delG mutation in the tail domain of keratin 5: a Japanese family with epidermolysis bullosa simplex with mottled pigmentation. J Invest Dermatol 2005;125:83–5.
33 Chan YMC, Jäckel A, Zabel B et al. A human keratin 14 ‘knockout’: the absence of K14 leads to severe epidermolysis bullosa simplex and a function for an intermediate filament protein. Genes Dev 1994;8:2574–87.
34 Rugg EL, McLean WHI, Lane EB et al. A functional ‘knockout’ of human keratin 14. Genes Dev 1994;8:2563–73.
35 Jonkman MF, Heeres K, Pas HH et al. Effects of keratin 14 ablation on the clinical and cellular phenotype in a kindred with recessive epidermolysis bullosa simplex. J Invest Dermatol 1996;107:764–9.
36 Corden LD, Mellerio JE, Gratian MJ et al. Homozygous nonsense mutation in helix 2 of K14 causes severe recessive epidermolysis bullosa simplex. Hum Mutat 1998;11:279–85.
37 Hovnanian A, Pollack E, Hilal L et al. A missense mutation in the rod domain of keratin 14 associated with recessive epidermolysis bullosa. Nature Genet 1993;3:327–32.
38 Koss-Harnes D, Høyheim B, Anton-Lamprecht I et al. A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: two identical de novo mutations. J Invest Dermatol 2002;118:87–93.
39 McGrath JA, Hoeger PH, Christiano AM et al. Skin fragility and hypohidrotic ectodermal dysplasia resulting from ablation of plakophilin 1. Br J Dermatol 1999;140:297–307.
Dystrophic Epidermolysis Bullosa
Definition.
This is a group of inherited disorders characterized by mechanically induced blistering occurring immediately below the lamina densa of the basement membrane zone. Because of the characteristic level of cleavage, DEB is sometimes termed dermolytic EB. These disorders derive the name ‘dystrophic’ from the tendency of the blisters to heal with scarring.
Aetiology and Pathogenesis.
Dystrophic epidermolysis bullosa may be inherited as an autosomal dominant or an autosomal recessive trait. In general, it tends to be most severe when inherited as a recessive trait and mildest when inherited as a dominant trait, but there is considerable clinical overlap. In sporadic cases, it is often impossible to determine the mode of inheritance on clinical grounds alone. In the majority of cases of dominant DEB, there is a clear family history, suggesting a low rate of new mutations. Most sporadic cases of DEB seem to be recessive, even if clinically mild, although de novo dominant mutations are well recognized. This should be borne in mind when offering genetic counselling to affected families, and clarification by mutation analysis sought if necessary and available.
There are few data to indicate the prevalence of DEB. In Norway, the prevalence of dominant DEB has been estimated to be 1.4 per million [1]. In England, an estimated prevalence of all recessive types of EB was 3 per million [2], of which most were probably cases of DEB. A more recent estimated prevalence from Scotland was 21.4 per million for all types of DEB [3]. In the USA, the estimated prevalence of dominant and recessive DEB is around 1 per million for each type, and about 0.5 per million for cases in which the inheritance pattern is unknown [4]. These figures suggest a carrier frequency for recessive DEB of about 1 in 350 [5].
Linkage and mutation analyses have demonstrated that all types of DEB result from mutations in the gene for type VII collagen (COL7A1) [6,7] on 3p21.1 [8]. Type VII collagen is the major component of anchoring fibrils [9,10] and is a large molecule (approximately 1000 kDa), which is synthesized and secreted by keratinocytes and fibroblasts. Structurally, it comprises a homotrimer of three α1 (VII) chains that associate to form a triple helix. The molecule has three domains: a central triple-helical domain consisting of (Gly-X-Y) repeats and two non-helical globular domains, termed NC-1 and NC-2. Type VII procollagen molecules associate via their carboxy termini, following which the NC-2 domains are cleaved off. The resulting type VII collagen molecules further condense to form anchoring fibrils.
It has become increasingly clear that the type of COL7A1 mutation or mutations is able to predict the clinical severity and prognosis of an individual’s DEB [11]. Specifically, most dominant DEB results from glycine substitution mutations in the collagenous triple-helical domain, causing dominant-negative interference between wild-type and mutant proteins and disruption of protein secretion and/or homotrimer formation [7,12–14]. In recessive disease, the severe generalized (Hallopeau–Siemens) form results from mutations that cause premature termination of translation on both COL7A1 alleles, resulting in little or no functional protein [11,15–17]. The milder forms of recessive disease, however, result from more diverse types of mutation, such as splice site [18], missense [19], silent glycine substitution [18,20] and delayed termination codon [21] mutations.
Pathology.
Dystrophic epidermolysis bullosa is the clinical reflection of defective attachment of the basement membrane to the underlying dermis, manifest at the ultrastructural level, in most cases, by reduced numbers of morphologically abnormal anchoring fibrils [22]. The monoclonal antibody LH-7.2 binds to the NC-1 domain of type VII collagen at the basement membrane zone in normal skin. Although there is generally no binding of this antibody at all in severe recessive DEB, binding is often weakly present in milder recessive DEB, but is usually normal in dominant DEB [23–27].
Clinical Features.
There is very wide variation in the severity of DEB in different patients, reflecting the many different mutations that may affect the type VII collagen gene. At its least severe, DEB can allow an almost normal quality and length of life, whereas at its most severe it may cause major handicap and a relatively brief and painful life.
Skin
The clinical hallmark of DEB is the tendency for blistered areas to heal with atrophic scarring and the development of contractures (Figs 118.11, 118.12). Although the presence of milia in recently healed areas is highly characteristic of dystrophic forms of EB, milia may be seen transiently in other types of EB, especially in neonates (Fig. 118.13).
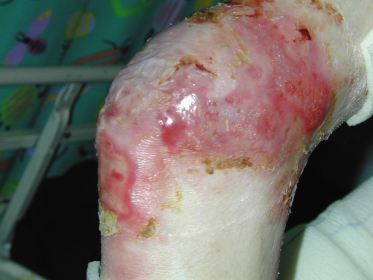
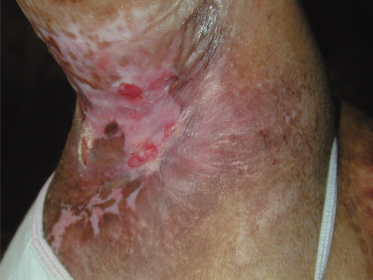
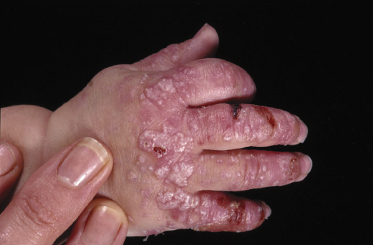
Dystrophic epidermolysis bullosa may be divided into a number of distinct subtypes, although, in practice, there may be a degree of overlap between them [28]. The major DEB subtypes comprise dominant DEB, severe generalized recessive DEB (formerly Hallopeau–Siemens) and recessive DEB generalized, other (formerly non-Hallopeau–Siemens). Other important but less common subtypes of DEB are discussed below.
Blistering in DEB tends to be provoked predominantly by knocks and blows to the skin rather than by rubbing, as is the case in EBS. Blisters in DEB therefore occur most frequently at skin sites where such trauma is common, such as the dorsa of the hands and feet, and the elbows and knees. However, persistent rubbing of the skin also predisposes to blistering, especially rubbing by clothing and bedding, particularly around the neck, waist, groins, hips and lumbosacral area. Another prominent cause of blistering is the attachment of anything adherent to the skin, such as adhesive tapes; this feature has great practical importance in the initial clinical recognition and management of the disease in the neonate.
The blisters may vary considerably in size, and may exceed 10 cm in diameter if left untreated. They tend to be flaccid and filled with either clear or blood-stained fluid. Healing of previously unblistered sites is generally fairly rapid. Crops of milia are common following initial re-epithelialization, but in areas not subject to further blistering the milia disappear after a few months. Discernible scarring is unusual following a single episode of blistering in a particular area, and generally only follows recurrent blistering. Blistering is much more easily provoked in areas that have previously been blistered, particularly when scarring has occurred. Healing areas tend to itch, and when the patient scratches further blistering is likely to follow. This can establish a cycle of blistering, itching and reblistering, the situation being made worse by the decreasing quality of healing and the increasing ease with which reblistering therefore occurs.
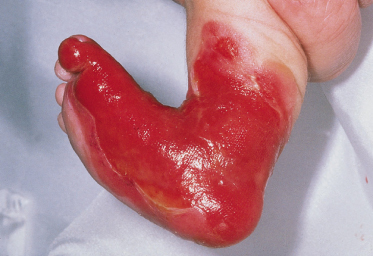
In the great majority of cases, blisters or, more often, erosions are present at or very shortly after birth. If the infant has been delivered by caesarean section, the presenting lesions may be deceptively mild, but this may not be an indication of the severity of the condition. Not uncommonly, an extensive eroded or ulcerated area is present at birth on one or both lower legs, usually on the dorsum and medial aspect of the foot and on the medial and anterior aspect of the shin (Fig. 118.14). This type of lesion almost certainly evolves in utero as a result of the fetus rubbing one leg against the other. When unilateral and in the absence of other lesions suggestive of EB, the significance of this type of lesion may not be appreciated. Such congenital absence of the skin on the lower leg is not specific for any particular type of EB [29]. These areas usually heal fairly rapidly, although the resulting scarring may lead to some deformity, particularly to upward displacement of the great toe and diminished growth of the foot.
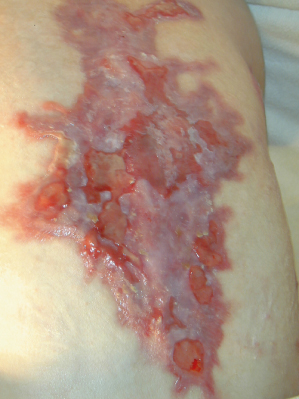
New blisters may develop less frequently with increasing age. It is unclear whether this is because of a genuinely decreased tendency of the skin to blister or is simply a reflection of more effective avoidance of trauma by older patients. In patients who are more severely affected, this trend is often counterbalanced by the steadily increasing fragility of skin that has been repeatedly ulcerated in the past, and which becomes atrophic and as delicate as tissue paper. Whereas previously ulcerated areas would usually heal rapidly, these atrophic areas may now break down so frequently that they never seem to heal. The neck, axillae, elbows, hands, hips, back, knees and ankles seem to be among the most troublesome sites from this point of view (Fig. 118.15). Most patients who survive into adult life may nevertheless enjoy a gradual improvement in the quality of scarred areas alongside a greater resistance of the skin to the development of new blisters.
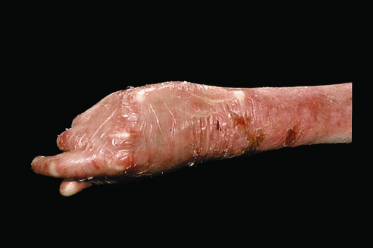
In those with mild DEB, scarring can almost disappear in adult life, leading to a remarkable improvement in the appearance of, for example, the hands. Patients with DEB quite frequently develop areas of dark macular pigmentation with rather characteristic irregular borders at sites of previous blisters. These so-called ‘EB naevi’, also seen in EBS and JEB, may look highly suspicious clinically and histologically, but have not been associated with, nor developed into, malignant melanoma [30]. Some patients with DEB develop ivory white papules that are most commonly seen on the trunk. These ‘albopapuloid’lesions were formerly felt to be specific for a type of dominant DEB (Pasini variant) [31] but they are now recognized to occur in other cases of dominant and recessive DEB.
The most sinister late complication of recessive, and to a much lesser extent dominant, DEB is the tendency for squamous cell carcinomas (SCCs) to develop in recurrently ulcerated and scarred areas, particularly over bony prominences on the limbs [32–35]. The incidence of these tumours reaches its peak in the third and fourth decades, but they may arise in early adolescence [36]. Clinical assessment of SCC may be difficult in these patients as appearances may be very similar to the chronic ulcerated areas typical of the more severe forms of EB. Areas that fail to heal or any hyperkeratotic nodules should be treated with suspicion and biopsied for histological analysis. Where an ulcer measures more than a few centimetres across, it may be necessary to biopsy at more than one site from the ulcer margin to ensure that any neoplastic change is not missed as a result of a sampling error. If patients are unaware of the danger of developing skin cancer they may fail to bring such lesions to medical attention in good time. However, even with prompt and appropriate surgical management of EB SCCs, patients tend to develop further, increasingly aggressive primary SCCs over time. As a result, metastatic disease is the major cause of death in patients with severe generalized recessive DEB [37]. The cumulative risk of developing SCC in this form of EB is 7.5% by age 20 years, 80.2% by 45 years, and 90.1% by 55 years [37] (compared with a risk of 9–14% for men and 4–9% in women from the non-EB general population in the USA [38]). An increased incidence of SCC is also well described in junctional EB, although the risk appears to be less than for patients with DEB [35,37,39].
Epidermolysis Bullosa Pruriginosa.
There is a fairly distinctive clinical variant of DEB in which the predominant features are pruriginous papules and/or lichenified plaques, associated with scarring [40,41]. Lesions occur mostly on the limbs, particularly on the shins, but may also be seen on the trunk. Itching is marked. Finger and toenail dystrophy is very common, with thickening or loss of the nail plate. Because intact blisters are rarely seen, the fact that the patient has DEB can easily be overlooked, and alternative diagnoses such as lichen planus, nodular prurigo and dermatitis artefacta may be considered. Mucosal lesions tend to be absent. Interestingly, the onset of clinical features is very variable, and may occur at any time between early childhood and the third decade. Where familial cases have occurred, inheritance has generally been autosomal dominant, although recessive and sporadic inheritance patterns are also well recognized [42]. From molecular analyses of these patients, there is no clear correlation between the site and nature of mutations in the type VII collagen gene and the expression of the pruriginosa phenotype, suggesting that additional factors may be responsible for developing this clinical subtype [42].
Pretibial Epidermolysis Bullosa [43–45].
In this variant of DEB, blistering and scarring are predominantly located on the shins. There may be associated toenail dystrophy. There may be overlap with EB pruriginosa in patients who experience significant pruritus as part of their disease.
Inversa Epidermolysis Bullosa [46,47].
In this unusual variant of DEB, skin fragility is mainly limited to the proximal flexures, whereas other sites such as the hands and feet are relatively spared. In addition to flexures, the mouth, oesophagus and external ear canals are particularly badly affected. The majority of cases of inversa DEB are recessively inherited, but there is no clear pattern of genotype-phenotype correlation and the reasons for this distinctive clinical pattern are not known.
Bullous Dermolysis of the Newborn [48–51].
A subgroup of infants with DEB can be defined on the basis of abundant amounts of type VII collagen found intraepidermally on immunocytochemistry that correspond to the presence of stellate bodies in basal keratinocytes ultrastructurally. This is thought to represent abnormal handling and secretion of type VII collagen within the epidermis. These infants have a relatively good prognosis, with most enjoying virtual resolution of their disease by the end of the first year of life, although initially blistering may be severe, even fatal [52]. Some individuals continue to experience blistering into childhood, however, so the formerly used term ‘transient’ bullous dermolysis of the newborn has now been changed [28]. The intraepidermal type VII collagen may disappear within weeks of birth, so that it is important to take skin biopsies as early as possible, although prolonged intraepidermal retention may persist for years after clinical resolution in some cases [53]. Although the majority of affected infants have autosomal dominant inheritance, recessive cases are recognized, and type VII collagen gene mutations may be identified in both types [53–55].
Hands
Cutaneous scarring in DEB may lead to a variety of complications, particularly to joint contractures and to fusion of the fingers and toes (Fig. 118.16). Progressive hand deformity is common in patients who blister readily. When digital fusion is going to develop, one can usually detect the earliest signs of the process within the first year of life. By this age, it is possible to gain some idea of the likely speed of its progression. The process of fusion seems to occur rather insidiously in the apices of the interdigital spaces, where careful examination will generally reveal small fissures. Occasionally, accidental trauma will lead to denudement of skin on the opposing aspects of more than one finger. If the fingers are opposed during healing, they may become fused within hours. Children with severe DEB are always at risk of acute loss of the entire skin cover of one or more fingers or of an entire hand, an injury known as ‘degloving’. This is especially likely to happen when a small child stumbles while holding an adult’s hand who then reacts by holding on tightly to prevent the child from falling, only to remove the skin from much of the hand.
Fig. 118.16 Severe recessive DEB, showing scarring, digital fusion and loss of nails and distal digits.
Surprisingly good hand function may be retained despite marked digital fusion, so long as apposition of index finger and thumb is retained. More disabling is the development of flexion contraction of the hand as a result of fibrosis of the skin on the palmar aspects of the fingers and hand.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


