Skin and hair/nail keratins are subdivided on the basis of their end domains and their charge: acidic (soft keratins 10–21 and hard keratins 31–38, type I) or basic/neutral (soft keratins 1–9 and hard keratins 81–86, type II). The acidic keratins are coded for by a gene cluster on chromosome 17, the basic ones by genes on chromosome 12. The two forms also differ slightly in their basic structure: type I keratins lack H1 and H2 domains [4].
Keratins exist as obligate heterodimers of type I and type II chains [5]. The association between the chains is thought to take place through the rod domain, which shows a motif of seven amino acids (the ‘heptad motif’), thought to mediate the association of the helices. Short, strongly conserved sequences at the beginning and end of the rod domains, the helix initiation and helix termination motifs, respectively, are essential for the initiation and proper termination of the dimerization process. Alterations in this motif perturb keratin function and are generally associated with severe skin disease. The keratin filaments aggregate into a network (Fig. 117.2), that interacts through specialized molecules called plakins [6] with adhesion structures in the keratinocytes: desmosomes and hemi-desmosomes [7]. Thus the cytoskeleton of each cell is linked to its membrane and, via this membrane, to other cells. Emerging evidence suggests that other motifs in keratins mediate interactions with various signalling pathways in the cell, thus implying the cytoskeleton in signal transduction.
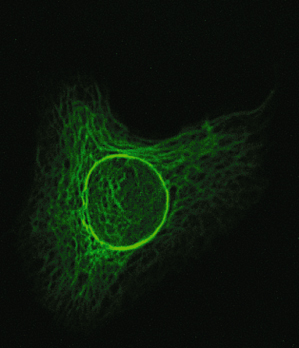
Fig. 117.2 HaCaT cell transfected with a green fluorescent protein-labelled keratin 1 construct. The intermediate filament network is clearly visible.
During differentiation of the epidermal cells, the expression of keratins in the keratinocytes changes as the cells move upwards (Table 117.1). Basal keratinocytes express K4 and K15. Upon their maturation to spinous layer cells, K5 and K14 are downregulated and K1, K2e and K10 are expressed [8]. Nail and hair progenitor cells express mainly K6, K16 and K17. These keratins are also expressed in other regions in response to keratinocyte stress or injury. Because of this, they can also be found in the palms and soles. K6a and K16 are also expressed in mucosal epithelia. K9 is expressed in the palms and soles only [9]. Non-epidermal keratins are expressed in mucous membranes and two of these, K4 and K13, have been implicated in a disorder of the mucosa (see below).
Table 117.1 Differentiation-specific expression of keratins
| Layer of epidermis | Expression of keratins |
| Stratum spinosum | |
| Upper | K2e |
| Lower | K1/K10, K6/K16, K4/K13, K9* |
| Stratum basale | K5/K14, K6b, K17 |
* Expression in palmoplantar skin only.
As the keratinocytes reach the stratum granulosum, the expression of keratins ceases. Among the many proteins produced in the upper layer are loricrin and filaggrin [10]. These two are a major part of the so-called cornified envelope (CE), an insoluble hydrophobic layer on the inner surface of the plasma membrane of the keratinocytes in the stratum corneum. This layer is made by cross-linking of the proteins mentioned, both to each other and to the KIF network [10]. A number of enzymes are involved in this process, mainly transglutaminases [10].
Keratins and other members of the IF family are involved in maintaining the structural integrity of the epidermis. Disruption of the IF network has been proposed to cause abnormal formation of the corneal layer and/or decreased resistance of the epidermis to mechanical stress. However, recent research is providing evidence to suggest that the diseases associated with keratin (and other IF) mutations do not result from epithelial fragility alone. For example, it has been shown that loss of keratin 17 correlates with decreased Akt/mTOR signalling activity. Furthermore, two amino acids in K17 are required to relocalize the adaptor protein 14-3-3-σ from the nucleus to the cytoplasm [2]. These findings reveal a new role for the cytoskeleton in regulating cell growth and metabolism. It is to be expected that further research will continue to deepen our insight in this manner.
Symptoms caused by keratin mutations may also be related to the affected cells’ response to the presence of misfolded (mutant) keratin proteins. Meanwhile, the number of disorders due to keratin mutations is growing and now includes liver and ocular disease, showing that keratins are also involved in the maintenance of simple epithelia (see, for example, reference [11]).
References
1 McLean WH, Irvine AD. Disorders of keratinisation: from rare to common genetic diseases of skin and other epithelial tissues. Ulster Med J 2007;76(2):72–82.
2 Kim S, Wong P, Coulombe P. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature 2006;441(7091):362–5.
3 Yu J, Yu DW, Checkla DM et al. Human hair keratins. J Invest Dermatol 1993;101(1 suppl):56S–9S.
4 Strelkov SV, Herrmann H, Aebi U. Molecular architecture of intermediate filaments. Bioessays 2003;25(3):243–51.
5 Herrmann H, Hesse M, Reichenzeller M et al. Functional complexity of intermediate filament cytoskeletons: from structure to assembly to gene ablation. Int Rev Cytol 2003;223:83–175.
6 Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res 2007;313(10):2189–203.
7 Holthofer B, Windoffer R, Troyanovsky S et al. Structure and function of desmosomes. Int Rev Cytol 2007;264:65–163.
8 Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell Biol 2008;129(6):705–33.
9 Knapp AC, Franke WW, Heid H et al. Cytokeratin No. 9, an epidermal type I keratin characteristic of a special program of keratinocyte differentiation displaying body site specificity. J Cell Biol 1986;103(2):657–67.
10 Steinert PM, Marekov LN. The proteins elafin, filaggrin, keratin intermediate filaments, loricrin, and small proline-rich proteins 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J Biol Chem 1995;270(30):17702–11.
11 Ku NO, Strnad P, Zhong BH et al. Keratins let liver live: mutations predispose to liver disease and crosslinking generates Mallory–Denk bodies. Hepatology 2007;46(5):1639–49.
Disorders Caused by Keratin Mutations
Known disorders caused by keratin mutations are listed in Table 117.2. These are discussed below. Note that several keratin diseases are grouped under other nosological headings and are discussed in more detail elsewhere. Therefore, this chapter will provide an overview of these disorders; for details the reader is referred to the appropriate chapters. Keratin gene mutations are catalogued in the Human Intermediate Filament database: www.interfil.org [1]. A note on terminology: keratin genes are indicated by ‘KRT’, the proteins by ‘K’.
Table 117.2 Keratin disorders
| Disorder (and subtypes) | Keratins affected |
| Epidermolysis bullosa, simplex type; Naegeli–Franchescetti–Jadassohn syndrome, dermatopathia pigmentosa reticularis | K5/14 |
| Dowling–Degos disease | K55 |
| Epidermolytic ichthyosis (cullous congenital ichthyosiform erythroderma of Brocq) | K1/10 |
| Epidermolytic epidermal naevus | K1/10 (mosaic) |
| Epidermolytic palmoplantar hyperkeratosis (Vörner) | K1/9 |
| Ichthyosis bullosa Siemens | K2e |
| Pachyonychia congenital | K6a/6b/16/17 |
| Focal NEPPK | K16 |
| Steatocystoma multiplex | K17/6b |
| White sponge naevus | K4/13 |
| Cryptogenic liver cirrhosis | K8/18 |
| Meesmann’s corneal dystrophy | K3/12 |
| Monilethrix | K83, 81, 86 |
| Ectodermal dysplasia, ‘pure’ hair-nail type | K85 |
| Pseudo-folliculitis barbae | K75 |
NEPPK, non-epidermolytic palmoplantar keratoderma.
Epidermolysis Bullosa Simplex (MIM #131760, #131800, #131900, KRT5/14)
The inherited forms of epidermolysis bullosa (EB) have recently been reclassified and EB simplex (EBS) is no longer subdivided into three entities (Weber–Cockayne, Dowling–Meara and Koebner) [2]. Nowadays, EBS is considered as a single entity with variable expressivity, which is caused by mutations in KRT5 and KRT14. The term ‘simplex’ refers to the level of blistering. EB with mottled pigmentation can be caused by mutations in KRT5/14 [3,4] and likewise is assigned to the EBS group. It is of interest to note here that there are some forms with associated symptoms that must be distinguished because they have a different molecular background. EBS with muscular dystrophy and/or pyloric atresia is caused by mutations in the PLEC1 gene coding for the intermediate filament-binding protein plectin [5]. Mutations in the ITGB4 gene coding for integrin-β4 have also been reported in EBS but are associated with nail dystrophy and enamel hypoplasia [6].
Both keratins 5 and 14 are expressed in basal epidermal cells. Different mutations can give rise to phenotypes of varying severity; there are also strong environmental influences such as ambient heat and humidity. It has been shown that disturbed network formation is an essential component of EBS [7]. Interestingly, a number of disorders formerly though to be distinct have now been shown to be allelic to EBS. From a nosological point of view, one might argue that they should perhaps no longer be recognized as separate disease entities. However, their clinical appearance is sufficiently distinctive and of mnemonic value to retain the separate categories. These are discussed below.
Naegeli–Franchescetti–Jadassohn Syndrome and Dermatopathia Pigmentosa Reticularis (MIM #161000, #125595, KRT14)
These disorders are discussed under the same heading because their clinical features are highly similar, if not identical, and because they are allelic [8]. In the authors’ opinion, they should probably be considered as manifestations of EBS. In their typical form, though, they are sufficiently distinct to be recognized as a defined phenotype.
Distinguishing symptoms of Naegeli–Franceschetti–Jadassohn syndrome (NFJS) are complete absence of dermatoglyphics (fingerprint lines), skin hyperpigmentation, palmoplantar keratoderma, reduced sweating, nail dystrophy and discoloured, yellow teeth that are lost early in life [9]. The hyperpigmentation is illustrated in Figure 117.3
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


