Cutis Marmorata
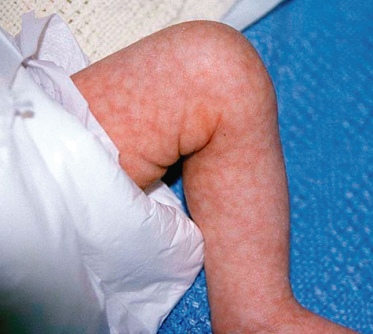
Cutis marmorata is a frequent finding in children with trisomies 21 and 18 and is also seen in the much rarer trisomy 10p. Trisomy 18 (Edwards syndrome) occurs in 1:6000 livebirths and is characterized by invariable in utero growth restriction, congenital heart disease, unusual hand posture, rocker-bottom feet and a high incidence of diaphragmatic hernia. Most die in utero or in the first few days of life but prolonged survival has been described [20]. Another chromosomal abnormality to consider in children with prominent cutis marmorata is del 7q, especially if associated with split-hand foot malformation [21]. Cutis marmorata has also been described in mosaic trisomy 4 [22] and del 12q [23] (Fig. 116.2).
Thin Skin
Thin skin is a feature of the sex chromosome aneuploidy disorder 48,XXYY. These patients are males with similar features to those with Klinefelter syndrome (47,XXY) in that they are tall, have a eunuchoid habitus, sparse body hair, small penis and testes and peripheral vascular disease leading to leg ulcers and varicosities (see below) [24]. Patients with 48,XXYY have developmental delay and a lower IQ than patients with Klinefelter syndrome.
Lipoatrophy
The most common chromosomal disorder in which generalized lipoatrophy can be seen is trisomy 18 (see above). Lipoatrophy has also been described in rarer disorders such as dup16q [25], del19q [26] and mosaic trisomy 3 [27], all uncommon.
Lymphoedema
Lymphoedema is frequently seen in Turner syndrome (45,XO) and can present as early as 12 weeks’ gestation where it manifests as increased nuchal translucency on ultrasound. Nuchal lymphoedema commonly resolves by birth but the webbed neck seen in patients with Turner syndrome is thought to be partly explained by the preceding in utero nuchal oedema [28]. Lympoedema involving the hands and feet occurs in approximately 80% of neonates with Turner syndrome and can persist, but usually resolves by age 2 [28]. Unilateral or bilateral lymphoedema may recur in childhood and adulthood. The pathogenesis of lymphoedema in Turner syndrome is aplasia or hypoplasia of the lymphatics and is pathologically distinct from the structurally abnormal lymphatics that cause nuchal oedema in Down syndrome, trisomy 13 and trisomy 18 [29]. Lymphoedema has also been described in numerous chromosomal disorders including trisomy 9 [30], dup 5q [31], del 7q [32], del 15q [33] and ring (X) [34].
Excessive/Premature Skin Wrinkling
Excessive skin wrinkling has been reported in the dup1q syndrome. Other notable features are macrocephaly, seizures and autistic features [35]. Premature wrinkling of the skin is a consistent feature of Down syndrome [10].
Hyperextensible Skin
Loose or hyperextensible skin is common in fragile X syndrome (see above) in association with soft skin and joint hypermobility. Hyperextensible skin is also seen in del 6q syndrome where it can resemble cutis laxa [36]. Other features of this syndrome are mental retardation, large ears and congenital heart disease. Sacral agenesis and imperforate anus have been described.
Hypohidrotic Ectodermal Dysplasia (HED) (see Chapter 127)
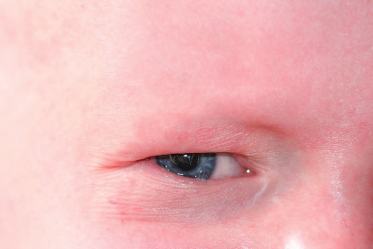
The discovery of the causative gene for X-linked HED (ectodysplasin-A on Xq12) came as a result of a case of HED in a woman with a translocation between the X chromosome and chromosome 9 (t(X;9)) which identified the region of the X chromosome where the gene resides [37]. Subsequent cases of X;autosome translocations have been reported involving the X chromosome and other autosomes, with the breakpoint always being close to Xq28 [38]. In addition to the classic features of hypohidrosis, hypotrichosis, spoon-shaped nails and hypodontia/conical teeth, children with HED due to a translocation could be predicted to have associated cognitive impairment if the translocation is unbalanced (Fig. 116.3).
Fig. 116.3 Absent eyebrows and fine periorbital wrinkling in hypohidrotic ectodermal dysplasia (HED).
Patchy Abnormalities of the Skin
Cutis Aplasia/Hypoplasia
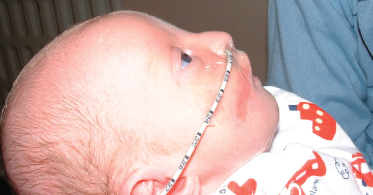
The chromosomal disorder classically associated with cutis aplasia is trisomy 13 (Patau syndrome). Trisomy 13 occurs in 1:12000 births and is usually lethal in the perinatal period although, like trisomy 18, prolonged survival has been described [39]. The main features of trisomy 13 are holoprosencephaly, microcephaly, severe cognitive impairment, cleft lip/palate, polydactyly, congenital heart disease and cutis aplasia of the cranial vertex. In some instances, a patch of cutis aplasia is visible on prenatal ultrasound and assists in making a clinical diagnosis of trisomy 13 when other suggestive features are present. Midline scalp defects are also a recognized feature of Wolf–Hirschorn syndrome (del 4p) [40]. These patients have a characteristic craniofacial profile that gives the impression of a ‘Greek warrior helmet’ (Fig. 116.4) and have associated microcephaly, mental retardation, short stature and other skin findings including ear tags/pits, skin dimples and abnormal teeth. Cutis aplasia has also been described in patients with del 19q [28] and dup1q [41].
Fig. 116.4 Typical facial features of Wolf–Hirschorn syndrome (del 4p) showing the classic ‘Greek warrior helmet’ profile.
Macules/Papules
Macules and papules are common in many chromosomal disorders. A form of miliaria rubra has been described in the del 3q27 syndrome [42]. Anophthalmia has also been associated with several cases of this deletion [43] and has now been shown to be caused by haploinsufficiency of the SOX2 gene consequent on the deletion. Several affected patients also have oesophageal atresia and the combination of features is known as Rogers syndrome [44]. Agenesis of the corpus callosum and mental retardation are usual.
Ulceration
A tendency to develop recurrent venous ulcers of the legs has been described in males with Klinefelter syndrome (47,XXY) and in patients with the similar 48,XXYY phenotype. The incidence of ulceration on retrospective data is 6–13%, at least 20 times greater than the general population [45]. Deep vein thrombosis and pulmonary embolism also occur more commonly in Klinefelter syndrome which suggests that venous stasis secondary to an underlying coagulopathy is the cause of the ulceration. Increased platelet aggregation secondary to elevated plasminogen activator inhibitor-1 has been implicated as a possible cause [46] but elevated factor VIII levels [47], heterozygous mutations in factor V Leiden and prothrombin [48] and homozygous mutations in MTHFR [49] have been reported in individual patients. Whatever the precise cause, it is likely that the hypoandrogenism/hyperoestrogenism seen in Klinefelter patients is a contributing factor.
Keloid Scar Formation
It has long been thought that patients with Turner syndrome (45,XO) have an increased risk of keloid scar formation but the largest review to date documented an incidence of only 1% [50]. It is likely that keloid scar formation is over-represented in Turner patients because their surgery is often in areas that are high risk for keloid scar formation (e.g. the head, neck and upper chest), and if only for this reason, the risk of keloid scar formation should be discussed. Keloids are a not uncommon finding among patients with Rubinstein–Taybi syndrome, most of which cases are due to mutation within the CBP locus on chromosome 16p, but about 15% of whom can be identified by a FISH positive deletion encompassing the CBP locus. The syndrome is readily recognized by the mental retardation, broad angulated thumbs and halluces and characteristic facial appearance with down-slanting palpebral fissures and prominent nasal columella.
Acromial Dimples
Bilateral acromial dimples are noteworthy features of the 18q deletion syndrome [51]. These dimples are seen along the superior margin of the scapulae and should raise suspicion of an underlying chromosomal disorder if other features are present. 18q deletion syndrome is characteristically associated with narrow external auditory canals.
Elastosis Perforans Serpiginosa (EPS)
Elastosis perforans serpiginosa is rare in childhood and is usually confined to patients with underlying connective tissue disorders (particularly Ehlers–Danlos type IV) but it is one of several skin conditions that occur more commonly in patients with Down syndrome (T21) [52].
Milia-Like Calcinosis Cutis
Milia-like calcinosis cutis is a rare benign condition primarily affecting the hands and feet. Two- thirds of cases occur in patients with Down syndrome but the reason for this is not known [53]. The mean age of presentation is 10 years.
Hyperkeratosis and Keratosis Pilaris
Palmoplantar hyperkeratosis and keratosis pilaris occur in approximately 10% of children with Down syndrome [54,55]. Keratosis pilaris has also been reported in the 18p deletion syndrome, a well-recognized syndrome in which mental retardation, holoprosencephaly, cleft lip/palate and marked slowness of motion are features. The widely spaced nipples, webbed neck, short stature and low posterior hairline have led to diagnostic confusion with Turner syndrome [56].
Abnormal Pigment
Diffuse Depigmentation
Diffuse depigmentation can be seen in Down syndrome but is also seen in Prader–Willi and Angelman syndromes which are due, in most cases, to parent-of-origin specific deletions of 15q11-13. Children with both conditions usually have fair skin and hair in addition to their dysmorphic and neurodevelopmental phenotypes. Prader–Willi syndrome can be caused by several genetic mechanisms involving chromosome 15 but in 70% of cases is due to a deletion at 15q11 on the paternally inherited chromosome. Prader–Willi patients typically present with neonatal hypotonia and poor feeding. After a period of difficult feeding and growth, they become hyperphagic in the second year of life and eventually obese. All have developmental delay and short stature. Angelman’s syndrome is caused by the identical deletion on the maternally inherited chromosome 15 and is characterized by severe speech and learning delay, ataxia and hand flapping behaviour. The hypopigmentation in both Prader–Willi and Angelman syndromes is thought to occur because the 15q11-13 deletion includes the OCA2 gene at 15q11.2, the gene for oculocutaneous albinism type II [57].
Patchy Depigmentation
Areas of patchy depigmentation should raise suspicion of an underlying chromosomal disorder when other features such as mental retardation are present. Generically these conditions used to be known as hypomelanosis of Ito, but that appellation has now been superseded by the more correct ‘Ito disease’ which recognizes the importance of this clinical sign as a marker of non-specific chromosomal mosaicism.
Patchy depigmentation has been specifically described in del 4q, where it is a helpful diagnostic feature [58]. The patchy depigmentation is thought to result from deletion of the c-kit proto-oncogene at 4q12.
Patchy depigmentation is a common finding in Pallister–Killian syndrome (mosaic tetrasomy 12p), described below.
Patchy Increased Pigmentation
Patchy increased skin pigmentation may also indicate an underlying chromosomal disorder, particularly mosaicism for a chromosomal disorder. A good example is Pallister–Killian syndrome (mosaic tetrasomy 12p), a syndrome in which mental retardation is commonly accompanied by diaphragmatic hernia, accessory nipples, frontotemporal balding, a sacral appendage, anorectal anomalies, polydactyly and patches of hyper- and hypopigmentation (Fig. 116.5). The pigmentary change develops after infancy and Wood’s light examination is sometimes required to appreciate the skin changes. Pallister–Killian syndrome results from tissue-limited mosaicism for two extra identical copies of the short arm of chromosome 12 (isochromosome 12p). This occurs as the result of a meiotic error in a parental gamete (usually maternal) which leaves the zygote with two normal chromosomes 12 plus the additional isochromosome 12p. After conception, some cells (particularly lymphocytes) lose the isochromosome and become diploid, thereby establishing mosaicism and making a standard cytogenetic diagnosis virtually impossible. The abnormal cell lines are usually found in fibroblasts and thus a karyotype performed on fibroblasts collected from a skin biopsy is the investigation of choice.
Other examples of hyperpigmentation in chromosomal mosaicism are mosaic trisomy 14 and mosaic trisomy 12. Complete trisomy 14 is universally fatal but when mosaicism occurs a well-recognized syndrome results. Patients have a distinct phenotype consisting of growth and psychomotor retardation, a broad nose, dysplastic low-set ears, micrognathia, congenital heart disease and patchy hyper- and hypopigmentation in most [59]. The pattern of pigmentation is similar in mosaic trisomy 12 but the general phenotype is poorly defined [60].
Pigmented Naevi
Pigmented naevi occur with increased frequency in numerous chromosomal disorders, the most common being Turner syndrome (45,XO and ring (X) [30]. Pigmented naevi can occur in any location and are seen in 50% of patients. Patients with several hundred naevi have been described [61]. The increased incidence of naevi does not translate to an increased risk of melanoma in Turner patients, raising the question of possible tumour suppressive factors at work [62]. The cause of the increased incidence of pigmented naevi in Turner syndrome patients is not known but treatment with human growth hormone or hormone replacement therapy, common in Turner patients, does not appear to play a causative role [28,63]. Multiple pigmented naevi can also occur in del Xq [64].
Café-au-lait Spots
Café-au-lait spots are commonly associated with chromosomal disorders and have been reported in the del 18p syndrome [65], Turner syndrome [28] and the Klinefelter variant 48,XXYY [24]. Café-au-lait spots are also frequently seen in chromosome breakage disorders, for example Fanconi syndrome and Bloom syndrome, both of which can also have more generalized areas of hyper- and hypopigmentation. In general, the presence of multiple café-au-lait spots in any child who is not considered to have neurofibromatosis type 1 but who has developmental delay or major congenital abnormalities should direct the investigations towards a possible chromosomal cause. Café-au-lait patches are an occasional finding in Silver–Russell syndrome cases but are probably no more likely to attend the isodisomy 7 cases of this phenotype than they are to signal the11p methylation abnormality-related phenotype [66,67].
Excessive Freckling
Excessive freckling has been reported in del 1p36 in association with other common features of the deletion syndrome, including a long narrow face, lax joints, mild hypertonia, developmental delay and ear asymmetry [68–70].
Vascular Abnormalities
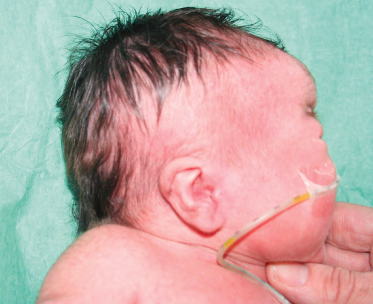
Telangiectasia
Telangiectasias are an important sign in the chromosome breakage disorder ataxia-telangiectasia (see chromosome breakage, part 1). The telangiectasias can be found anywhere but tend to occur in the lateral margins of the eyes and on the surface of the cheeks and external ear. Telangiectasias can be the physical sign that leads to the diagnosis (Fig. 116.6).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


