X-linked recessive traits exclusively occur in male individuals. Characteristically, one generation is skipped because the disease is transmitted by clinically healthy female carriers. X-linked dominant non-lethal traits occur in both sexes. By contrast, X-linked dominant male-lethal traits are observed exclusively in female individuals because the underlying mutation has a lethal effect on hemizygous male embryos. Exceptionally, such traits may also occur in male individuals with a normal XY constitution and this can be explained by an early postzygotic mutation or by a gametic half-chromatid mutation [23]. These men can transmit the trait to their daughters [24]. Conversely, a mother-to-son transmission of such phenotypes is not possible and has never been observed, although this has been erroneously stated in the literature [25,26].
Y-Linkage
In Y-linked traits, two different modes of transmission should be distinguished. First, there are pseudo-autosomal genes that have their counterpart on the X chromosome [27]. The pseudo-autosomal region at the end of the short arm of the Y chromosome undergoes crossing over with the telomeric part of the X chromosome. Traits determined by such genes show a transmission similar to autosomal traits. Second, the remaining Y-linked genes can be transmitted from father to son only [27].
Some examples of Y-linked traits are summarized in Table 115.4. A Y-linked gene relevant to paediatric dermatology is the SRY gene controlling the testis-determining factor localized at Yp11.3 [28]. Girls affected with MIDAS syndrome (microphthalmia–dermal aplasia–sclerocornea) may show anomalies of external or internal genitalia and may display an SRY-positive status, indicating a translocation between the pseudo-autosomal parts of Xp and Yp [29].
Table 115.4 Y-linked traits
| Trait | MIM number |
| Stature/tooth size | 475 000 |
| Testes-determining factor (SRY) | 480 000 |
| Interleukin 3 receptor, Y-linked | 430 000 |
| Granulocyte–macrophage colony-stimulating factor receptor, α-subunit, Y-linked | 425 000 |
| Hairy pinnae of the ear (?) | 425 500 |
SRY, sex-determining region.
Hairy pinnae of the ear is another phenotype that may be Y-linked [30]. The trait is relevant to dermatology but certainly not to paediatric dermatology.
Exceptions to and Variations on Mendelian Inheritance
Today, so many exceptions to the rules of Mendelian inheritance have been brought to light that it is astonishing that Mendel was able to formulate the rules that have immortalized his name. Some of these modifying concepts and mechanisms are reviewed below.
Mitochondrial Inheritance
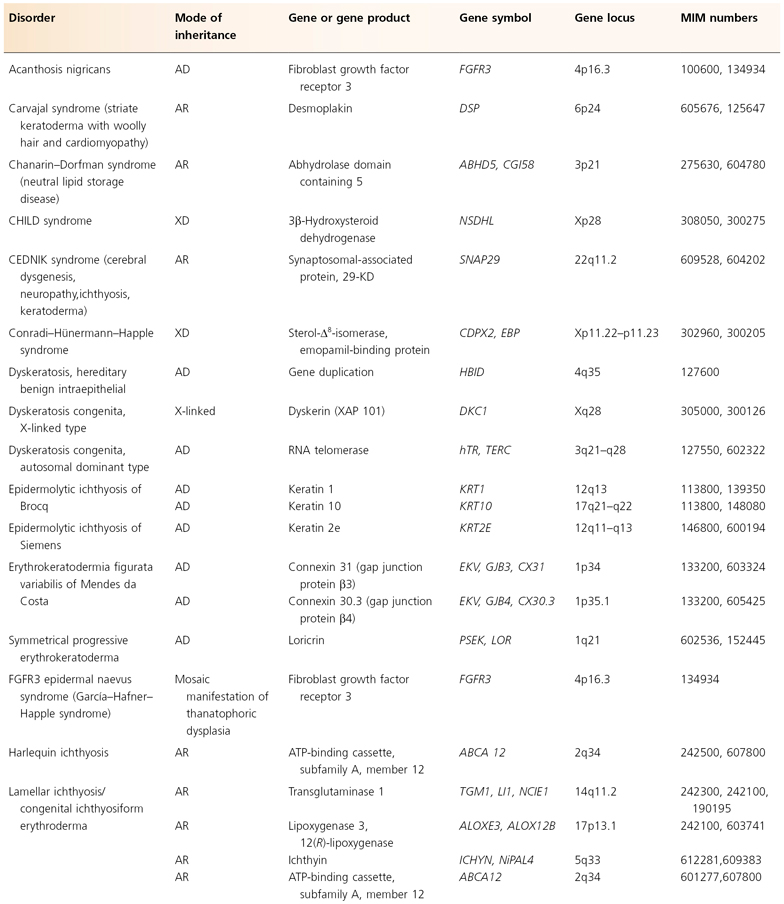
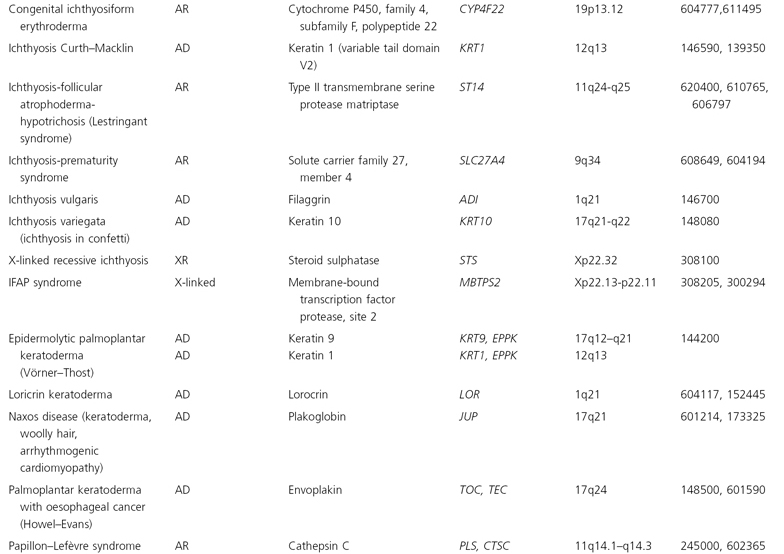
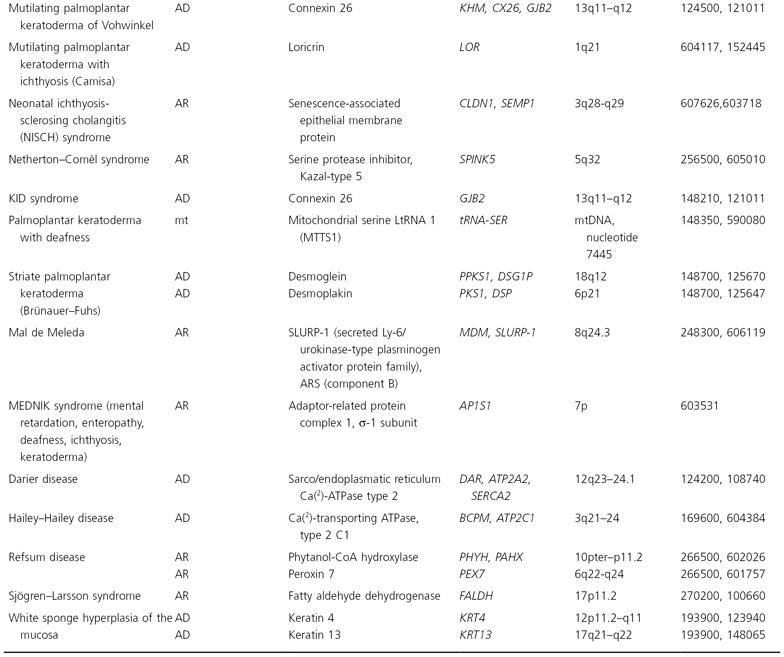
The human mitochondrial genome contains 14 protein-coding regions that have been completely sequenced [31]. They are contained in a circular mitochondrial chromosome resembling that of a bacterium. There are no introns and there is very little flanking non-coding DNA. The mitochondrial mode of transmission is exclusively matrilineal. The presently known catalogue of mitochondrial disorders contains one skin disorder in the form of palmoplantar keratoderma with deafness (MIM 590080; see Table 115.5) [32].
Table 115.5 Hereditary disorders of keratinization
AD, autosomal dominant; AR, autosomal recessive; mt, mitochondrial.
Genomic Imprinting
Some genes show the phenomenon of imprinting. When such a gene is derived from the mother, it acts in a different way from that derived from the father [33]. This different expression is apparently due to a different degree of methylation of DNA during gametogenesis [34]. Imprinting has been proposed to be present in neurofibromatosis I [35] and familial glomus tumours [36] and it may also play a role in the inheritance of atopy [37] and psoriasis [38].
Anticipation
Many doctors believed in anticipation more than 100 years ago. A genetic trait should worsen progressively from generation to generation. Until recently, modern geneticists have regarded this idea as a superstition. Remarkably, however, the concept of anticipation has now experienced a revival. In some traits, such as fragile X syndrome, myotonic dystrophy or Huntington’s chorea, the number of trinucleotide repeats may increase from generation to generation, correlating with an increasing severity of the disease [39,40]. Anticipation has also been assumed in autosomal dominant dyskeratosis congenita [41] and Behçet disease [42,43].
Uniparental Disomy
A child suffering from an autosomal recessive disorder may have only one heterozygous parent. Molecular research has shown that, by way of exception, both of the mutant chromosomes may be derived from one parent [41,42]. Uniparental disomy has been documented in various recessive skin disorders [44] (Box 115.1).
Box 115.1 Examples of uniparental disomy causing autosomal recessive skin disorders
- Recessive dystrophic epidermolysis bullosa [45]
- Junctional epidermolysis bullosa of the Herlitz type [46]
- Congenital insensitivity to pain with anhidrosis (CIPA) [47]
- Netherton syndrome [48]
Compound Heterozygosity
An individual may carry two dissimilar mutant alleles at the same locus. For example, compound heterozygosity for different mutations in COL7A1 gives rise to a moderately severe dystrophic epidermolosysis bullosa phenotype [49]. Compound heterozygosity plays an important role in many other autosomal recessive skin disorders such as mal de Meleda [50], congenital erythropoietic porphyria [51], congenital ichthyosiform erythroderma [52], xeroderma pigmentosum [53] or pseudo-xanthoma elasticum [54]. It should not be confused with double heterozygosity, which means heterozygosity at each of two separate loci on the same chromosome.
Semi-Dominant Inheritance
This term is rather loosely applied when heterozygotes show a very mild phenotype, whereas homozygotes or compound heterozygotes are more severely involved, as noted in ichthyosis vulgaris [55]. In principle, all semi-dominant disorders can also be categorized as dominant traits.
Pseudo-Dominance
A seemingly dominant transmission of a recessive gene may occur when a clinically affected individual is mating with a heterozygous gene carrier. In this particular situation, the children have a 50% risk of recurrence. This may happen in a population with a high degree of inbreeding. Such pedigrees have been observed in mal de Meleda, a distinctive type of palmoplantar keratoderma occurring predominantly in the population of the island of Mljet (Meleda) off the Croatian coast [56]. Another example of pseudo-dominance is pseudo-xanthoma elasticum that is today categorized exclusively as a recessive trait [54].
Paradominance
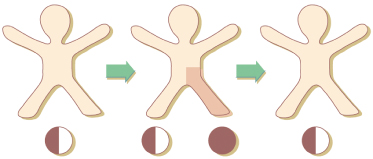
Some mosaic phenotypes such as Becker naevus or Sturge–Weber–Klippel–Trenaunay syndrome usually occur sporadically but may affect, by way of exception, several members of a family. To explain this paradox, the concept of paradominant transmission has been proposed [57,58]. Heterozygous gene carriers would be, as a rule, phenotypically normal and for this reason, the mutant gene would be transmitted imperceptibly through many generations. The trait would become manifest only when a somatic event of allelic loss in an early stage of embryogenesis gave rise to a clone of cells that were either homozygous or hemizygous for the mutation (Fig. 115.2). In several other mosaic traits not fitting an established mode of Mendelian inheritance, the concept of paradominant transmission (see below) may replace the terminological vagueness of ‘incomplete penetrance’ or ‘reduced expression’.
Fig. 115.2 Paradominant inheritance. Heterozygotes are phenotypically normal. The trait is expressed only when loss of heterozygosity occurs in an early stage of embryogenesis and gives rise to a clonal population of cells, either homozygous or hemizygous for the mutation.
Gonadal Mosaicism
An autosomal dominant trait may occur in several siblings although both parents are clinically healthy. This phenomenon has been conclusively explained by gonadal mosaicism [59]. Molecular proof of this concept has been provided in many hereditary disorders such as tuberous sclerosis [60], bullous ichthyosis of Brocq [61], neurofibromatosis type 1 [62] and epidermolysis bullosa simplex [63]. Gonadal mosaicism may be due to an early postzygotic mutation affecting both germ cells and somatic tissues, or to a late postzygotic mutation affecting the germ cells alone. Such events should no longer be called ‘somatic mutations’. Only those postzygotic mutations affecting somatic cells alone, without any germline mosaicism, are true somatic mutations (Fig. 115.3). Similarly, gonadal mosaicism for an X-linked trait, incontinentia pigmenti, has been documented in a healthy male [64].
Fig. 115.3 A postzygotic mutation may involve both somatic and germinal tissues (left), somatic tissue only (middle) or germinal tissue only (right). The term ‘somatic mutation’ exclusively refers to a postzygotic event as shown in the middle panel.

The concept of gonadal mosaicism is important for genetic counselling. In a sporadic case of a severe autosomal dominant disorder, molecular analysis of the father’s sperm may be a reasonable approach to better characterize the risk of gonadal mosaicism, especially when the mutation rate of the involved gene is relatively high, as in neurofibromatosis type 1 [62].
Founding Effect
In an isolated population originating from a limited number of founding parents, some autosomal recessive disorders may be either absent or, conversely, particularly frequent due to the absence or presence of the mutation in the founding parents. For example, cartilage–hair hypoplasia is relatively common in the Finnish-speaking population of Finland, whereas phenylketonuria is virtually absent [65].
Point Mutations Versus Deletions
Many traits showing a monogenic mode of inheritance are due not to a point mutation but to a deletion either present within a given gene or involving several neighbouring genes. It should be noted that the human genes have different sizes. For example, the insulin gene is relatively small [66] whereas the steroid sulphatase gene is quite large [67]. This may explain the rather high frequency of deletions found within the steroid sulphatase gene [68].
Contiguous Gene Syndromes
Large deletions may involve several neighbouring genes, giving rise to a ‘contiguous gene syndrome’. For example, a large deletion at Xp22.3 may result in a combination of X-linked recessive ichthyosis, chondrodysplasia punctata of the Maroteaux type and Kallmann syndrome (hypogonadotrophic hypogonadism with anosmia) [69]. Other examples are the contiguous gene syndromes of tuberous sclerosis 2 and polycystic kidney disiease [70] or hypotrichosis simplex and holoprosencephaly [71]. In such cases, the different entities are inherited together, simulating a monogenic multisystem birth defect.
Genetic Heterogeneity
Molecular research has provided evidence that similar phenotypes are often genetically different. For example, two types of tuberous sclerosis are so far indistinguishable at the clinical level. The underlying genes have been assigned to chromosomes 9q and 16p [8]. Other examples of genetic heterogeneity are xeroderma pigmentosum and Waardenburg syndrome (see below). The term should not be applied to polygenic traits.
Hypomorphic Alleles
Mutations of the X-linked NEMO (IKK-γ) gene usually give rise to incontinentia pigmenti that represents a male-lethal phenotype. Some ‘hypomorphic’ alleles (that is, those with a lower level of gene expression) within the same gene, however, may cause a non-lethal trait in the form of ectodermal dysplasia of Zonana (hypohidrotic ectodermal dysplasia with immune deficiency) [4]. In this trait, female carriers do not suffer from classic incontinentia pigmenti, although they show a pigmentary pattern following the lines of Blaschko [5]. Another example is the fact that hypomorphic EBP alleles cause a peculiar syndrome in male individuals, whereas other EBP mutations give rise to an X-linked dominant, male-lethal trait in the form of Conradi–Hünermann–Happle syndrome [72].
Epigenetic Inheritance
In mice, dogs and other animals, variegated coat patterns may occur as a non-Mendelian hereditary trait. For example, the murine mutant agouti viable yellow (Avy) shows a great variability of expression, reflecting differences in the epigenetic state [73]. Both variegation and variable expressivity are caused by the action of a retrotransposon. Transposable elements or retrotransposons are particles of retroviral origin that are interspersed in large amounts in the mammalian genome [74]. During early embryogenesis, retrotransposons may silence or activate the expression of neighbouring genes by methylation or demethylation [75]. Some familial cases of human pigmentary mosaicism have tentatively been explained by the action of such metastable epialleles (see below) [76].
Polygenic Inheritance
Many hereditary skin diseases are determined by more than one gene. Such polygenic traits are psoriasis [77], atopic dermatitis [78] and multiple melanocytic naevi, including dysplastic naevi [79,80]. In polygenic traits, several scientific terms, such as expression, penetrance, pleiotropism and genetic heterogeneity, are meaningless and should be avoided. In all of these traits, impressive pedigrees suggesting a dominant mode of inheritance have been documented but can be explained by a bias of ascertainment. Such phenotypes should no longer be taken as ‘autosomal dominant traits with reduced penetrance and variable expression’.
Polygenic Versus Multifactorial Traits
These two terms are often used synonymously but the term ‘polygenic’ is preferred because a ‘multifactorial trait’ implies the influence of environmental factors too.
In polygenic traits, the risk of recurrence is not constant but increases with the number of affected individuals present in a given family. When compared with monogenic traits, it is rather difficult to elucidate the molecular basis of polygenic skin disorders [77,81]. However, important advances have been made in the genetic understanding of such common skin diseases by documenting a close association of atopy with loci on several chromosomes [82], including the filaggrin locus at 1q21 [83], or of psoriasis with many other loci [77,84], including 8p23 showing a significant association between higher genomic copy numbers for β-defensin genes and risk of psoriasis [85]. It should be noted, however, that ‘linkage’ of such loci may vary to a high degree in different populations.
In classic genetics, twin studies have been very useful in elucidating polygenic inheritance. The principle is to study unselected twin pairs and to compare monozygotic to dizygotic pairs. In polygenic traits, a concordant manifestation is found in both types of twin pairs but tends to be much higher (four- to fivefold) in monozygotic twins [78]. The Minnesota study [86] has yielded important information regarding the influence of the environment on atopic manifestations. The total immunoglobulin E (IgE) levels of monozygotic pairs showed a remarkable concordance, even in those twins who had been separated at an early age and lived under different environmental conditions.
References
1 Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007;44:181–92.
2 Gruis NA, Bavinck JNB, Steijlen PM et al. Genetic linkage between the collagen VII (Co17A1) gene and the autosomal dominant form of dystrophic epidermolysis bullosa in two Dutch kindreds. J Invest Dermatol 1992;99:528–30.
3 Hovnanian A, Duquesnoy P, Blanchet–Bardon C et al. Genetic linkage of recessive dystrophic epidermolysis bullosa to the type VII collagen gene. J Clin Invest 1992;90:1032–6.
4 Zonana J, Elder ME, Schneider LC et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 2000;67:1555–62.
5 Kosaki K, Shimasaki N, Fukushima H et al. Female patient showing hypohidrotic ectodermal dysplasia and immunodeficiency (HED-ID). Am J Hum Genet 2001;69:664–6.
6 Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol 2007;6:340–51.
7 European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993;75:1305–15.
8 Napolioni V, Curatolo P. Genetics and molecular biology of tuberous sclerosis complex. Curr Genom 2008;9:475–87.
9 Méndez M, Poblete-Gutiérrez P, García-Bravo M et al. Molecular heterogeneity of familial porphyria cutanea tarda in Spain: characterization of 10 novel mutations in the UROD gene. Br J Dermatol 2007;157:501–7.
10 Cicardi M, Igarashi T, Rosen FS et al. Molecular basis for the deficiency of complement 1 inhibitor in type I hereditary angioneurotic edema. J Clin Invest 1987;79:698–702.
11 Quastel M, Harrison R, Cicardi M et al. Behavior in vivo of normal and dysfunctional C1 inhibitor in normal subjects and patients with hereditary angio-neurotic edema. J Clin Invest 1983;71:1041–6.
12 Nishisho I, Nakamura Y, Miyoshi Y et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991;253:665–9.
13 Shanley S, Ratcliffe J, Hockey A et al. Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet 1994;50:282–90.
14 Oetting WS, King RA. Molecular analysis of type I-A (tyrosine negative) oculo-cutaneous albinism. Hum Genet 1992;90:258–62.
15 Herman ML, Farasat S, Steinbach PJ et al. Transglutaminase-1 gene mutations in autosomal recessive congenital ichthyosis: summary of mutations (including 23 novel) and modeling of TGase-1. Hum Mutat 2009;30:537–47.
16 Huber M, Rettler I, Bernasconi K et al. Mutations of keratinocytic transglutaminase in lamellar ichthyosis. Science 1995;267:525–8.
17 Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961;190:372–3.
18 Happle R. X-chromosome inactivation: role in skin disease expression. Acta Paediatr 2006;95(451)(suppl):16–23.
19 Lyon MF. The Lyon and the LINE hypothesis. Semin Cell Dev Biol 2003;14:313–18.
20 Happle R. Lyonization and the lines of Blaschko. Hum Genet 1985;70:200–6.
21 Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005;434:400–4.
22 Happle R. Cutaneous manifestation of X-linked genes escaping inactivation. Clin Exp Dermatol 1992;17:69.
23 Bornholdt D, Oeffner F, König A et al. PORCN mutations in focal dermal hypoplasia: coping with lethality. Hum Mutat 2009;30:E618–28.
24 Mahé A, Couturier J, Mathé C et al. Minimal focal dermal hypoplasia in a man: a case of father-to-daughter transmission. J Am Acad Dermatol 1991;25:879–81.
25 Kurczynski TW, Berns JS, Johnson WE. Studies of a family with incontinentia pigmenti variably expressed in both sexes. J Med Genet 1982;19:447–51.
26 Hecht F, Hecht BK, Austin WJ. Incontinentia pigmenti in Arizona Indians including transmission from mother to son inconsistent with the half chromatid mutation model. Clin Genet 1982;21:293–6.
27 Burgoyne PS. Genetic homology and crossing over in the X and Y chromosomes of mammals. Hum Genet 1982;61:85–90.
28 Jäger RJ, Harley VR, Pfeiffer RA et al. A familial mutation in the testis determining gene SRY shared by both sexes. Hum Genet 1992;90:350–5.
29 Mücke J, Hepffner W, Thamm B et al. MIDAS syndrome (microphthalmia, dermal aplasia and sclerocornea): an autonomous entity with linear skin defects within the spectrum of focal hypoplasias. Eur J Dermatol 1995;5:197–203.
30 Stern C, Centerwall WR, Sarkar SS. New data on the problem of Y-linkage of hairy pinnae. Am J Hum Genet 1964;16:455–71.
31 Gilkerson RW. Mitochondrial DNA nucleoids determine mitochondrial genetics and dysfunction. Int J Biochem Cell Biol 2009;41:1899–906.
32 Online Mendelian Inheritance in Man (OMIM). www.ncbi.nlm.nih.gov/omim/.
33 Marx JL. A parent’s sex may affect gene expression. Science 1988;239:352–3.
34 Hall JG. Genomic imprinting: review and relevance to human diseases. Am J Hum Genet 1990;46:857–73.
35 Miller M, Hall JG. Possible maternal effect on severity of neurofibromatosis. Lancet 1978;ii:1071–3.
36 Van der Mey AGL, Maaswinkel-Mooy PD, Cornelisse CJ. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet 1989;ii:1291–4.
37 Cookson WOCM, Young RP, Sandford AJ et al. Maternal inheritance of atopic IgE responsiveness on chromosome 11q. Lancet 1992;340:381–4.
38 Traupe H, van Gurp PJM, Happle R et al. Psoriasis vulgaris, fetal growth and genomic imprinting. Am J Med Genet 1992;42:649–54.
39 Sutherland GR, Richards RI. Invited editorial. Anticipation legitimized: unstable DNA to the rescue. Am J Hum Genet 1992;51:7.
40 Harper PS, Harley HG, Reardon W et al. Anticipation in myotonic dystrophy: new light on an old problem. Am J Hum Genet 1992;51:10–16.
41 Armanios M, Chen JL, Chang YP et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA 2005;102:15960–4.
42 Fietta P.Behçet’s disease: familial clustering and immunogenetics. Clin Exp Rheumatol 2005;23(suppl 38):S96–105.
43 Panicker JN, Vinayan KP, Ahsan Moosa NV, Elango EM, Kumar AA. Juvenile Behçet’s disease: highlighting neuropsychiatric manifestations and putative genetic mechanisms. Clin Neurol Neurosurg 2007;109:436–8.
44 Spence JE, Perciaccante RG, Greig GM et al. Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet 1988;42:217–26.
45 Fassihi H, Lu L, Wessagowit V et al. Complete maternal isodisomy of chromosome 3 in a child with recessive dystrophic epidermolysis bullosa but no other phenotypic abnormalities. J Invest Dermatol 2006;126:2039–43.
46 Castori M, Floriddia G, Pisaneschi E et al. Complete maternal isodisomy causing reduction to homozygosity for a novel LAMB3 mutation in Herlitz junctional epidermolysis bullosa. J Dermatol Sci 2008;51:58–61.
47 Miura Y, Hiura M, Torigoe K et al. Complete paternal uniparental isodisomy for chromosome 1 revealed by mutation analyses of the TRKA (NTRK1) gene encoding a receptor tyrosine kinase for nerve growth factor in a patient with congenital insensitivity to pain with anhidrosis. Hum Genet 2000;107:205–9.
48 Lin SP, Huang SY, Tu ME et al. Netherton syndrome: mutation analysis of two Taiwanese families. Arch Dermatol Res 2007;299:145–50.
49 Shimizu H, Hammami-Hauasli N, Hatta N, Nishikawa T, Bruckner-Tuderman L. Compound heterozygosity for silent and dominant glycine substitution mutations in COL7A1 leads to a marked transient intracytoplasmic retention of procollagen VII and a moderately severe dystrophic epidermolysis bullosa phenotype. J Invest Dermatol 1999;113:419–21.
50 Nellen RG, van Geel M, Steijlen PM, van Steensel MA. Compound heterozygosity for ARS component B mutations in a Dutch patient with mal de Meleda. Br J Dermatol 2009;160:878–80.
51 Kang TW, Oh SW, Kim MR, Lee JS, Kim SC. Compound heterozygosity for a premature termination codon and missense mutation in the exon 10 of the uroporphyrinogen III cosynthase gene causes a severe phenotype of congenital erythropoietic porphyria. J Eur Acad Dermatol Venereol 2009;23:470–1.
52 Akiyama M, Sakai K, Hatamochi A, Yamazaki S, McMillan JR, Shimizu H. Novel compound heterozygous nonsense and missense ABCA12 mutations lead to nonbullous congenital ichthyosiform erythroderma. Br J Dermatol 2008;158:864–7.
53 Fujimoto M, Leech SN, Theron T et al. Two new XPD patients compound heterozygous for the same mutation demonstrate diverse clinical features. J Invest Dermatol 2005;125:86–92.
54 Ringpfeil F, McGuigan K, Fuchsel L et al. Pseudoxanthoma elasticum is a recessive disease characterized by compound heterozygosity. J Invest Dermatol 2006;126:782–6.
55 Sandilands A, O’Regan GM, Liao H et al. Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. J Invest Dermatol 2006;126:1770–5.
56 Eckl KM, Stevens HP, Lestringant GG et al. Mal de Meleda (MDM) caused by mutations in the gene for SLURP-1 in patients from Germany, Turkey, Palestine and the United Arab Emirates. Hum Genet 2003;112:50–6.
57 Happle R. Paradominant inheritance: a possible explanation for Becker’s pigmented hairy nevus. Eur J Dermatol 1992;2:39–40.
58 Happle R. Klippel–Trenaunay syndrome: is it a paradominant trait? Br J Dermatol 1993;128:465.
59 Hall JG. Somatic mosaicism: observations related to clinical genetics. Am J Hum Genet 1988;43:355–63.
60 Verhoef S, Vrtel R, van Essen T et al. Somatic mosaicism and clinical variation in tuberous sclerosis complex. Lancet 1995;345:202.
61 Paller AS, Syder AJ, Chan YM et al. Genetic and clinical mosaicism in a type of epidermal nevus. N Engl J Med 1994;331:1408–15.
62 Lázaro C, Ravella A, Gaona A et al. Neurofibromatosis type I due to germline mosaicism in a clinically normal father. N Engl J Med 1994;331:1403–7.
63 Nagao-Watanabe M, Fukao T, Matsui E et al. Identification of somatic and germline mosaicism for a keratin 5 mutation in epidermolysis bullosa simplex in a family of which the proband was previously regarded as a sporadic case. Clin Genet 2004;66:236–8.
64 Kirchman TTT, Levy ML, Lewis RA et al. Gonadal mosaicism for incontinentia pigmenti in a healthy male. J Med Genet 1995;32:887–90.
65 Vogel F, Motulsky AG. Human Genetics: Problems and Approaches, 2nd edn. Berlin: Springer Verlag, 1986: 509–11.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


