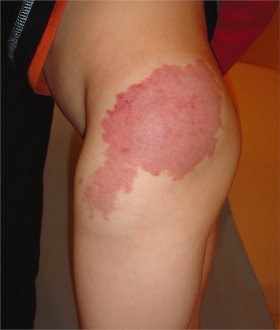
Puberty and trauma can trigger expansion as fast-flow becomes clinically evident. A purple colour and a mass develop. Other local signs include warmth, a thrill, a bruit and pulses of increased amplitude (Schobinger Stage I). The most common location is head and neck (70%) [12]. As an AVM worsens, draining veins become prominent, tortuous and distended (Schobinger Stage II) (see Fig. 112.1). Wherever the site of an AVM, the eventual consequences are darkening of skin colour, recurrent ulceration, pain and intermittent bleeding (Schobinger Stage III) [12–14]. These changes can occur in childhood, and haemorrhage can be life-threatening. A facial AVM localized to the skin and/or bones (ethmoid, maxilla or mandible) can cause asymmetrical hypertrophy and gingival bleeding [14]. A nasal AVM can cause epistaxis. AVM in a digit gradually causes ischaemic cutaneous changes and shortening of the nail and distal phalanx. Pseudo-Kaposi sarcomatous cutaneous changes occur in association with AVM in the lower limb [15], but rarely before adolescence. Cardiac failure can be the end-stage of a large AVM (Schobinger Stage IV).
Radiological investigation is recommended whenever AVM is suspected clinically [13]. A number of techniques define the nature and extent of the lesion.
- Ultrasonography combined with grey-scale and colour Doppler. An AVM is heterogeneously echogenic and exhibits low-resistance, high-velocity arterial flow and pulsatile venous flow [13]. Vessels are tortuous. Pulsed Doppler measures the arterial output (of carotid, axillary or femoral arteries) compared with the uninvolved side, and allows serial evaluation in a non-invasive fashion.
- Computed tomography (CT) with contrast demonstrates the extent of an AVM but, unlike magnetic resonance imaging (MRI), cannot definitely differentiate between haemangioma, venous malformation (VM) and AVM. Three-D angio-scan may be useful to analyse the feeding and draining vessels portraying the AVM, especially in childhood.
- MRI. Flow-voids correspond to fast-flow vessels, in all sequences with gadolinium (spin-echo T1- and T2-weighted sequences) except gradient-echo (in which sequence increased signal is present in vascular spaces).
- Magnetic resonance angiography (MRA) portrays the anomalous vascular network. It replaces arteriography for follow-up of AVM.
- Arteriography depicts the angio-architecture of an AVM, especially prior to embolization.
Prognosis.
Arteriovenous malformation is the most unpredictable of all vascular malformations. Usually AVMs enlarge, in time, and cause local destruction. Once the initial diagnostic work-up is completed, the child should have periodic clinical evaluation and serial Doppler and/or MRI and MRA. An AVM usually remains quiescent in childhood. Failure to recognize its true nature can lead to dangerous mismanagement and complications. Partial excision and ligation of arterial feeders usually trigger AVM expansion, especially during puberty. Mismanagement of a limb AVM can lead to gangrene, necessitating amputation [16]. A large AVM, usually in a limb, can cause congestive heart failure, but rarely in childhood.
Differential Diagnosis.
In an infant or child, AVM must be distinguished from haemangioma (Fig. 112.2). Ultrasonography and colour Doppler demonstrate that haemangiomas have equatorial feeding arteries, peripheral veins, variable echogenicity and fast-flow, but no true arteriovenous (AV) shunting.
A few other dermatological lesions, uncommon in the paediatric patient, mimic AVM. Epithelioid haemangio-endothelioma usually occurs on the distal extremity as a purple and locally aggressive lesion [17]. Lupus erythematosus tumidus or sarcoidosis on the ear or nose, and Melkersson–Rosenthal syndrome of the lip can masquerade as AVM. The pseudo-Kaposi sarcomatous changes, developing unilaterally in a severe AVM of the leg, are easily differentiated from true Kaposi sarcoma. Dabska tumour, in childhood, can simulate an AVM, when located in the ear.
Treatment.
In the paediatric age group, AVM is often clinically inconspicuous, but every AVM is potentially dangerous. Embolic and/or surgical treatment of a quiescent AVM is controversial. An AVM is usually not treated in its quiescent stage. The management of an AVM should be multidisciplinary, modest about predicting outcome, and should never overestimate therapeutic possibility. The only acceptable surgical stratagem is complete resection [14,16,18,19]. Partial excision leads to transient improvement, but the AVM inevitably re-expands over time. Proximal ligation or embolization of arterial feeding vessels is contraindicated; after a period of transient benefit, vascular recruitment occurs, as neighbouring arteries supply the nidus. Furthermore, arterial ligation prevents access to the nidus at a time when therapeutic embolization becomes necessary [20,21]. In a limb AVM, elastic stockings can provide some benefit and protect the skin from injury. Treatment of an AVM is complex and difficult. Intervention becomes necessary whenever local and/or cardiac complications occur.
A pharmacological approach using marimastat, a matrix metalloproteinase inhibitor, was successfully used to treat an extensive AVM of the arm that caused rapid progressive bony destruction in a young girl despite ethanol embolization. The treatment, done over a 9-year-period, resulted in improvement of the AV shunts and regrowth of bones. No adverse effects were reported [22].
Superselective Arterial Embolization
The most common embolic materials are liquid (ethanol, isobutylcyanoacrylate), particles (Ivalon) or implantable devices (coils, microspheres). Embolization alone as a palliative treatment is only indicated for complicated AVM (e.g. in case of ulceration or haemorrhage), when surgical excision will be disfiguring or mutilating. Embolization of an AVM, unlike embolization of a single direct AVF, is rarely ‘curative’. The aim of the treatment is to occlude the shunts with a permanent or an irritating material, such as histoacryl glue or coils, or ethanol; particles are not indicated. The site of injection of the embolic agent must be as close as possible to the nidus. Proximal occlusion causes secondary refilling of the nidus through collateral arteries. In patients with maxillary or mandibular AVM, embolization should be performed prior to dental extraction, to minimize haemorrhage. Microcatheter guidewire systems, as used in interventional neuroradiology, permit a superselective approach to the nidus [23]. Direct puncture of the nidus, in association with local arterial and venous compression, is necessary when arteries are tortuous, or in patients who have previously had arterial ligation.
Surgical Resection
Arterial embolization is usually performed prior to resection [23–25], to occlude the nidus and minimize bleeding during the procedure. It does not affect the margins of resection. Usually, the AVM and the overlying skin are widely excised. Microsurgical free-flap transfer may be necessary for reconstruction [24]. Cutaneous expansion, prior to embolization and resection, is an alternative surgical strategy. The overlying skin can be saved only if normal; vascular-stained skin left in place often leads to recurrence. Lytic bony lesions, for example in the mandible, can cause life-threatening haemorrhage requiring endovascular management [26], sometimes followed by resection. AVM in the distal extremity often recurs after technically successful embolization and a symptom-free interval [27,28]. Because AVM is so difficult to eradicate, the word ‘control’ rather than ‘cure’ should be used and patients must be followed for many years.
Syndromic Arteriovenous Malformation
Parkes Weber Syndrome (OMIM 608355)
Parkes Weber syndrome is an eponym used for capillary–arteriovenous malformation (CAVM) or capillary–arteriovenolymphatic malformation (CAVLM), with limb overgrowth. Usually the lower extremity is involved, sometimes the upper limb. In association with multifocal capillary malformations (CMs), they are caused by RASA1 mutations and are part of the CM-AVM phenotype (see below).
Cutaneous warmth and bruit are present as a result of numerous AV microfistulas along the affected limb, particularly near the joints. In early childhood, arteriography may show only diffuse hypervascularity of the limb, and AV fistulas become obvious later in life. Bony overgrowth may reach excess length of more than 5 cm. Associated lymphoedema causes major disability [29].
Management.
The child with fast-flow complex/combined vascular malformation in a limb is evaluated annually. Leg length is measured clinically during infancy. After the age of 2 years, leg length is evaluated radiologically and, if abnormal, this assessment is repeated yearly. A shoe-lift is provided on the normal side if leg length discrepancy is over 1.5 cm. Ultrasonography and grey-scale and colour Doppler evaluation of limb arterial and venous vessels should be performed when the child is 3 or 4 years old. Plain radiography detects skeletal changes. MRI, MRA and arteriography are necessary in evaluation of arterial feeders or draining veins penetrating muscles and bones in symptomatic patients.
Prognosis.
Common complications during puberty are limb overgrowth, lytic bony lesions, pathological fractures, painful skin ulceration, distal pseudo-Kaposi sarcoma skin changes, high cardiac output and congestive heart failure.
Treatment.
Management is fundamentally conservative. Elastic compression stockings provide relief of pain due to venous engorgement. In the presence of leg length discrepancy, an adapted shoe-lift prevents compensatory tilting of the pelvis. In cases of increased vascularization of the growth zone, embolization of the knee cartilage artery may retard excess growth. Stapling epiphyseodesis can, in our experience, stimulate the AVM of this syndrome. In contrast, percutaneous epiphyseodesis is less aggressive. Lengthening of the normal leg is another option, when the predicted final height of the child is poor; however, lengthening of a normal extremity has complications, and the risk/benefit ratio must be carefully pondered.
Bonnet–Dechaume–Blanc and Wyburn–Mason Syndromes
These eponyms denote AVM in centrofacial and/or hemifacial skin, the eye and the mesodiencephalon [30,31]. Retinal involvement defines Bonnet–Dechaume–Blanc syndrome, also known as Wyburn–Mason syndrome, although it is not universal. Brégeat syndrome has no retinal or choroidal AVM, but exhibits vascular anomalies in the conjunctiva. The skin lesions are red, warm and thick, rarely following a trigeminal distribution (as in Sturge–Weber syndrome). Ipsilateral retinal, optic nerve, chiasma, optic pathway, basal ganglia and cerebral AVMs can be present. The jaws, nose and mouth are sometimes involved. AVM is present at birth and progressively worsens. Epistaxis, exophthalmos and hemianopia may occur. Some patients manifest a variety of neurological symptoms, including intracranial haemorrhage from the brain AVM, as well as mental changes. Modern ophthalmological and neuroradiological tools, including MRA, are used to delineate the oculo-orbital and cerebral anomalies. Therapeutic possibilities are limited. Mismanagement of the superficial lesions (arterial or venous ligation, proximal embolization, partial excision, laser treatments) causes expansion. Treatment of orbital location is a challenge because of the anatomical and haemorrhagic characteristics of AVM in this location [32]. Neurosurgical procedures are rarely curative.
Cobb Syndrome
This sporadic syndrome is composed of cutaneous AVM, masquerading as capillary malformation, and spinal cord AVMs in the same metamere. The spinal cord AVM is either (a) intramedullary or perimedullary and fed by spinal cord arteries or (b) dural and fed by radicular meningeal arteries [33]. AVM in the vertebrae and paraspinal muscles can also occur in the same metamere. Patients are at risk for local complications such as limb hypertrophy and ulceration. Neurological complications, often beginning in childhood, include pain, sensory disturbance and neurogenic bladder and bowel; motor symptoms (monoplegia, paraplegia or quadriplegia) depend on the location and extent of the lesion. MRI is the best screening test and should be performed before selective angiography. The neurosurgical goal is complete removal of the spinal cord AVM nidus whenever possible. Neuroendovascular treatment is an alternative to surgical resection.
We could postulate that Cobb syndrome is part of the CM-AVM phenotype; however, it is not known if these patients have distant multiple CMs [34].
Inherited Arteriovenous Malformation
Rendu–Osler–Weber Disease (Hereditary Haemorrhagic Telangiectasia; HHT) (OMIM 187300)
Definition.
This autosomal dominant disorder has a prevalence of at least 1/5–8000 and with high penetrance without sex preponderance [35,36]. HHT is characterized by cutaneous and mucosal telangiectasia and visceral AVMs located in the liver, brain or spinal cord, and/or lungs. It can cause both recurrent bleeding from visceral telangiectasia and life- or function-threatening haemorrhages from visceral arteriovenous anomalies [37].
Pathology.
Various genotypes correspond to slightly different phenotypes of HHT. HHT-1, caused by loss-of-function mutation in endoglin, a transforming growth factor (TGF-β) receptor located on chromosome 9q34–1, is often associated with a higher incidence of pulmonary and cerebral AVMs than HHT-2 [38,39]. HHT-2 is caused by loss-of-function mutations in another TGF-β receptor called ALK-1, mapped to 12q11–14 [40]. Another gene (SMAD4) mapped on chromosome 18q21.1 has been identified in juvenile polyposis-haemorrhagic telangiectasia (JP-HT) which is characterized by early-onset intestinal polyps followed by telangiectasias and epistaxis [41]. At least one more low-frequency locus (HHT-3) exists, mapped to 5 q [39].
Clinical Features.
Telangiectases are punctate, linear, stellate or nodular, and occur on the cheeks, nose, lips, tongue and other oral mucous membranes, and fingers. Recurrent epistaxis, the most frequent symptom of HHT, occurs in 90% of patients and usually develops during late adolescence. Gastrointestinal bleeding, the second most common haemorrhagic manifestation, begins in adulthood and affects one-quarter of patients.
Patients with pulmonary disease can have dyspnoea or haemoptysis, and can die from massive haemoptysis or haemothorax. Pulmonary AVMs place one-half of these patients at risk for neurological complications from septic emboli, with brain abscesses. As a result of pulmonary AV shunting, a defect in the bacterial filtering function of the pulmonary circulation allows bacteria to enter the systemic circulation. Infection in a lung AVF can also result in bacteraemia.
Central nervous system AVM in HHT can cause acute headache and spinal cord or intracerebral haemorrhage, sometimes leading to significant cognitive and motor impairment [42]. Nasal, gastric or colonic endoscopy localizes bleeding sites [43].
The main differential diagnosis is CM-AVM (see below) and HBT, a benign hereditary telangiectasia not associated with AVM.
Treatment.
Electrodesiccation, laser (Nd : YAG, diode, carbon dioxide or argon lasers) sclerotherapy and embolization help stop bleeding. Patients with anaemia require iron replacement and, occasionally, blood transfusion. In patients with HHT and pulmonary AVMs, antibacterial prophylaxis is recommended for dental, respiratory, GI or genitourinary procedures that place the patient at risk for bacteraemia. Endovascular treatment of AVMs should be considered.
Investigations in families with HHT can detect asymptomatic, unruptured intracranial AVMs [42,44–47]. Early genetic diagnosis allows detection in a given affected family of those children requiring investigations, follow-up and treatment whenever feasible, if visceral AVMs are diagnosed [48]. This molecular diagnosis is recommended in newborns, as lethal cerebral haemorrhage from undiagnosed AVMs has been reported in such families [42]. Treatment guidelines must balance the lifetime risk for bleeding against surgical morbidity and mortality. The incidence of cerebrovascular anomalies, including micro-AVMs and cerebral telangiectasia, was considered to be 20%.
Capillary Malformation-Arteriovenous Malformation (CM-AVM) (OMIM 608354)
Definition.
Capillary malformation-arteriovenous malformation is a newly recognized disorder which has a prevalence of at least 1/100,000. This autosomal dominant disorder manifests as multiple small pinkish-red, slightly tan round-to-oval CMs [49–52].
Clinical Features.
Some CMs are present at birth, whereas others appear later, but remain asymptomatic throughout life. Their size varies from a few millimetres to several centimetres in diameter. They often have a pale halo and an increased flow by hand-held Doppler examination (Fig. 112.3). There is a large inter- and intrafamilial phenotypic variability. AVM is present in 30% of CM-AVM patients, 80% of which includes vein of Galen aneurysmal malformation and intracranial AVF. They are often symptomatic at birth or in early childhood. In contrast to HHT, visceral AVMs are rare; extracranial lesions rather affect skin, subcutis and sometimes muscles and bones and are located in the head and neck region. Parkes Weber syndrome associated with distant cutaneous CMs is also a phenotype [51].
Pathology.
Molecular genetic evaluation led to the identification of loss-of-function mutations in the RASA1 gene (OMIM 139150). RASA1 is a GTPase, an intracellular molecule implicated in the RAS pathway. It converts the active GTP-bound RAS into its inactive GDP-bound form [51–53].
Treatment.
Brain MRI and MRA should be performed on each CM-AVM infant as early symptomatic AVM can occur. In contrast to sporadic CM, pulsed-dye laser is rarely effective in CM of CM-AVM. The main differential diagnosis is HHT (see above).
References
1 Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg 1982;69(3):412–22.
2 Mulliken JB, Young AE. Vascular Birthmarks: Hemangiomas and Malformations. Philadelphia: WB Saunders, 1988.
3 Enjolras O, Riché MC, Mulliken JB, Merland J. Atlas des Hémangiomes et Malformations Vasculaires Superficielles. Paris: McGraw-Hill, 1990.
4 Takahashi K, Mulliken JB, Kozakewich HP, Rogers RA, Folkman J, Ezekowitz RA. Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest 1994;93(6):2357–64.
5 Esterly NB. Cutaneous hemangiomas, vascular stains and malformations, and associated syndromes. Curr Prob Pediatr 1996;26(1):3–39.
6 Enjolras O, Mulliken JB. The current management of vascular birthmarks. Pediatr Dermatol 1993;10(4):311–13.
7 Enjolras O, Mulliken JB. Vascular tumors and vascular malformations (new issues). Adv Dermatol 1997;13:375–423.
8 Trop I, Dubois J, Guibaud L et al. Soft-tissue venous malformations in pediatric and young adult patients: diagnosis with Doppler US. Radiology 1999;212(3):841–5.
9 Burrows PE, Laor T, Paltiel H, Robertson RL. Diagnostic imaging in the evaluation of vascular birthmarks. Dermatol Clin 1998;16(3):455–88.
10 Limaye N, Boon LM, Vikkula M. From germline towards somatic mutations in the pathophysiology of vascular anomalies. Hum Mol Genet 2009;18(R1):R65–74.
11 Brouillard P, Vikkula M. Genetic causes of vascular malformations. Hum Mol Genet 2007;16(2):R140–9.
12 Enjolras O, Logeart I, Gelbert F et al. [Arteriovenous malformations: a study of 200 cases]. Ann Dermatol Venereol 2000;127(1):17–22.
13 Paltiel HJ, Burrows PE, Kozakewich HP, Zurakowski D, Mulliken JB. Soft-tissue vascular anomalies: utility of US for diagnosis. Radiology 2000;214(3):747–54.
14 Kohout MP, Hansen M, Pribaz JJ, Mulliken JB. Arteriovenous malformations of the head and neck: natural history and management. Plast Reconstr Surg 1998;102(3):643–54.
15 Larralde M, Gonzalez V, Marietti R, Nussembaum D, Peirano M, Schroh R. Pseudo-Kaposi sarcoma with arteriovenous malformation. Pediatr Dermatol 2001;18(4):325–7.
16 McClinton MA. Tumors and aneurysms of the upper extremity. Hand Clin 1993;9(1):151–69.
17 Roudier-Pujol C, Enjolras O, Lacronique J et al. [Multifocal epithelioid hemangioendothelioma with partial remission after interferon alfa-2a treatment]. Ann Dermatol Venereol 1994;121(12):898–904.
18 Enjolras O, Borsik M, Herbreteau D, Merland JJ. [Management of arteriovenous malformations]. Ann Dermatol Venereol 1994;121(1):59–64.
19 Wu JK, Bisdorff A, Gelbert F, Enjolras O, Burrows PE, Mulliken JB. Auricular arteriovenous malformation: evaluation, management, and outcome. Plast Reconstr Surg 2005;115(4):985–95.
20 Coldwell DM, Stokes KR, Yakes WF. Embolotherapy: agents, clinical applications, and techniques. Radiographics 1994;14(3):623–43; quiz 45–6.
21 Yakes WF, Rossi P, Odink H. How I do it. Arteriovenous malformation management. Cardiovasc Intervent Radiol 1996;19(2):65–71.
22 Burrows PE, Mulliken JB, Fishman SJ, Klement GL, Folkman J. Pharmacological treatment of a diffuse arteriovenous malformation of the upper extremity in a child. J Craniofac Surg 2009;20 Suppl 1:597–602.
23 Burrows PE, Fellows KE. Techniques for management of pediatric vascular anomalies. In: Cope C, (ed) Current Techniques in Interventional Radiology. Philadelphia: Current Medicine, 1995, pp.12–27.
24 Dompmartin A, Labbe D, Barrellier MT, Theron J. Use of a regulating flap in the treatment of a large arteriovenous malformation of the scalp. Br J Plast Surg 1998;51(7):561–3.
25 Enjolras O, Deffrennes D, Borsik M, Diner P, Laurian C. [Vascular ‘tumors’ and the rules of their surgical management]. Ann Chir Plast Esthet 1998;43(4):455–89.
26 Benndorf G, Campi A, Hell B, Holzle F, Lund J, Bier J. Endovascular management of a bleeding mandibular arteriovenous malformation by transfemoral venous embolization with NBCA. AJNR 2001;22(2):359–62.
27 Sofocleous CT, Rosen RJ, Raskin K, Fioole B, Hofstee DJ. Congenital vascular malformations in the hand and forearm. J Endovasc Ther 2001;8(5):484–94.
28 White RI Jr, Pollak J, Persing J, Henderson KJ, Thomson JG, Burdge CM. Long-term outcome of embolotherapy and surgery for high-flow extremity arteriovenous malformations. J Vasc Interv Radiol 2000;11(10):1285–95.
29 Enjolras O, Chapot R, Merland JJ. Vascular anomalies and the growth of limbs: a review. J Pediatr Orthop B 2004;13(6):349–57.
30 Brégeat P. Brégeat syndrome. In: Vinken PJ, Bruyn GW (eds) Handbook of Clinical Neurology: The Phakomatoses. Amsterdam: North Holland, 1975, pp.474–9.
31 Bhattacharya JJ, Luo CB, Suh DC, Alvarez H, Rodesch G, Lasjaunias P. Wyburn–Mason or Bonnet–Dechaume–Blanc as cerebrofacial arteriovenous metameric syndromes (CAMS). A new concept and a new classification. Intervent Neuroradiol 2001;7:5–17.
32 Jiarakongmun P, Alvarez A, Rodesch G, Lasjaunias P. Clinical course and angioarchitecture of cerebrofacial arteriovenous metameric syndromes: three demonstrative cases and literature review. Intervent Neuroradiol 2002;8(3):251–64.
33 Hodes JE, Merland JJ, Casasco A, Houdart E, Reizine D. Spinal vascular malformations: endovascular therapy. Neurosurg Clin North Am 1994;5(3):497–509.
34 Thiex R, Mulliken JB, Revencu N et al. A novel association between RASA1 mutations and spinal arteriovenous anomalies. Am J Neuroradiol 2010;31(4):775–9.
35 Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006;43(2):97–110.
36 Shovlin CL, Sodhi V, McCarthy A, Lasjaunias P, Jackson JE, Sheppard MN. Estimates of maternal risks of pregnancy for women with hereditary haemorrhagic telangiectasia (Osler–Weber–Rendu syndrome): suggested approach for obstetric services. Br J Obstet Gynaecol 2008;115(9):1108–15.
37 Ference BA, Shannon TM, White RI Jr, Zawin M, Burdge CM. Life-threatening pulmonary hemorrhage with pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia. Chest 1994;106(5):1387–90.
38 McAllister KA, Grogg KM, Johnson DW et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet 1994;8(4):345–51.
39 Shovlin CL, Hughes JM, Tuddenham EG et al. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat Genet 1994;6(2):205–9.
40 Johnson DW, Berg JN, Baldwin MA et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet 1996;13(2):189–95.
41 Gallione CJ, Repetto GM, Legius E et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004;363(9412):852–9.
42 Morgan T, McDonald J, Anderson C et al. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu syndrome). Pediatrics 2002;109(1):E12.
43 Sabba C, Pasculli G, Lenato GM et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost 2007;5(6):1149–57.
44 ter Berg JW, Dippel DW, Habbema JD, Westermann CJ, Tulleken CA, Willemse J. Unruptured intracranial arteriovenous malformations with hereditary haemorrhagic telangiectasia. Neurosurgical treatment or not? Acta Neurochirurg 1993;121(1–2):34–42.
45 Kadoya C, Momota Y, Ikegami Y, Urasaki E, Wada S, Yokota A. Central nervous system arteriovenous malformations with hereditary hemorrhagic telangiectasia: report of a family with three cases. Surg Neurol 1994;42(3):234–9.
46 Matsubara S, Mandzia JL, ter Brugge K, Willinsky RA, Faughnan ME. Angiographic and clinical characteristics of patients with cerebral arteriovenous malformations associated with hereditary hemorrhagic telangiectasia. AJNR 2000;21(6):1016–20.
47 Maher CO, Piepgras DG, Brown RD Jr, Friedman JA, Pollock BE. Cerebrovascular manifestations in 321 cases of hereditary hemorrhagic telangiectasia. Stroke 2001;32(4):877–82.
48 Haneen S, Johanna H, Ulrich G et al. Mutation analysis of ‘Endoglin’ and ‘Activin receptor-like kinase’ genes in German patients with hereditary hemorrhagic telangiectasia and the value of rapid genotyping using an allele-specific PCR-technique. BMC Med Genet 2009;10:53.
49 Eerola I, Plate KH, Spiegel R, Boon LM, Mulliken JB, Vikkula M. KRIT1 is mutated in hyperkeratotic cutaneous capillary–venous malformation associated with cerebral capillary malformation. Hum Mol Genet 2000;9(9):1351–5.
50 Eerola I, Boon LM, Watanabe S, Grynberg H, Mulliken JB, Vikkula M. Locus for susceptibility for familial capillary malformation (‘port-wine stain’) maps to 5 q. Eur J Hum Genet 2002;10(6):375–80.
51 Revencu N, Boon LM, Mulliken JB et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat 2008;29(7):959–65.
52 Boon LM, Mulliken JB, Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev 2005;15(3):265–9.
53 Eerola I, Boon LM, Mulliken JB et al. Capillary malformation–arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet 2003;73(6):1240–9.
Slow-Flow Vascular Malformations
Venous Malformation
Localized or Extensive Venous Malformation
Definition and Aetiology.
Venous malformations (VMs) involve the collecting side of the vascular network. Most lesions are sporadic and unifocal (VM, 93%); 1% are multifocal. VMs can be localized or extensive and may be located in the head and neck (the most common location), extremities and/or trunk. VMs occur in skin, mucous membranes, various soft tissues and viscera, and even bones. The lesions are present at birth, or they become clinically evident later. Familial forms include cutaneomucosal venous malformation (VMCM, 1%) and glomuvenous malformation (GVM, 5%) [1–3]. Small, spot-like VMs and larger mixed capillary-venous malformations occur in some patients with hereditary cerebral cavernous malformations caused by mutations in the KRIT1 gene (CCM1) [4–6].
Pathology.
Venous malformation are composed of ectatic, poorly defined channels, interconnecting to form a complex network that permeates normal tissues. Some lesions contain only distorted, enlarged venous channels (erroneously labelled as ‘cavernous haemangioma’). Other lesions consist of a spongy combination of ectatic venous channels intermingled with capillaries. The lining endothelium is flat and quiescent. Walls are thin and irregularly lined by a discontinuous layer of smooth muscle cells, positive for smooth muscle α-actin [7]. Thrombosis and calcifications (phleboliths) are frequent. The abnormal venous network drains to adjacent veins, many of which are varicose and lack valves. Recent discoveries have identified somatic mutations in the TIE2 gene, encoding a receptor specific for endothelial cells in nearly 50% of sporadic VMs [8].
Clinical Features.
Ectatic venous channels within the dermis give the lesions their characteristic blue colour. VMs swell with exertion and when the region is dependent. Deformation of involved tissues slowly worsens. Skin temperature is normal. There is no thrill or bruit. Depending on the size and site of their lesion, patients complain of swelling, pain or a burning sensation. Increased stiffness and pain upon awakening are frequent as well as painful thrombosis and phleboliths.
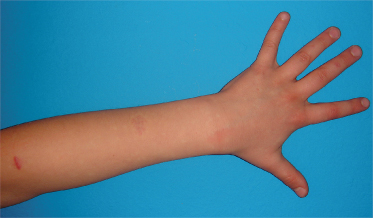
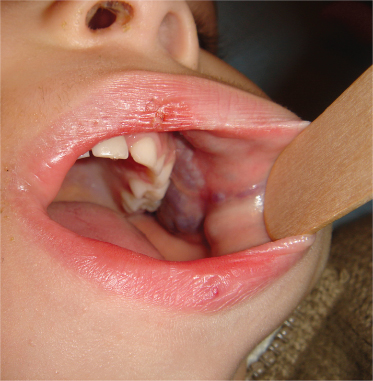
Head and neck VMs are usually unilateral; bilateral involvement occurs rarely. Owing to a mass effect, VMs can cause facial asymmetry, progressive distortion of features, dental malalignment and malocclusion (Fig. 112.4). Intraorbital VM varies in size, depending on head position. Enophthalmia can occur when the patient is standing, and minor exophthalmia when lying supine, but vision is not altered. Optic nerve compression rarely results from a VM in the orbital apex. Mucosal oral VMs involve the tongue, cheek or palate; they can extend to the parotid, parapharyngeal and laryngeal areas. VM in the oropharynx rarely impairs speech, but pharyngeal and laryngeal VMs can cause difficulties in swallowing, airway compromise and obstructive sleep apnoea.
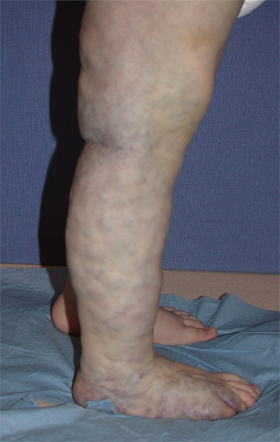
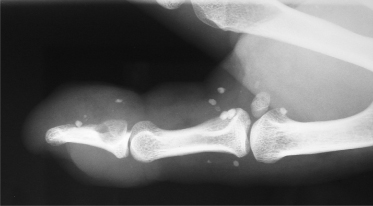
A pure VM in an extremity commonly invades muscles and joints (Fig. 112.5) [9–11]. Distal upper or lower limb VMs cause enlarged blue fingers or toes with sagging skin. VM affecting an entire upper or lower limb and adjacent trunk causes increased limb girth, but length discrepancy is uncommon. Genitalia, in both males and females, are commonly involved when there is lower extremity VM. VM slowly worsens in childhood and adolescence, and phleboliths develop. Amyotrophy is a common, early finding in a child with extensive limb VM; slight limb undergrowth is common. Slight overgrowth can also occur, but never to the extent seen in Klippel–Trenaunay syndrome. Weakening of the bony shaft caused by intraosseous VM may result in pathological fracture after minimal trauma [9].
Fig. 112.5 Extensive VM of lower extremity involving skin, subcutis, muscle and joint and associated with severe LIC.
Venous malformations exhibit a continuous cycle of thrombosis and thrombolysis, causing the formation of phleboliths (Fig. 112.6). Deep venous thrombosis is uncommon and pulmonary embolism is extremely rare. The VM-associated chronic localized intravascular coagulopathy (LIC) is characterized by elevated D-dimer level (>0.5 µg/mL) in 40% of patients [9,12]. LIC is associated with large size deep involvement of underlying tissues and presence of phleboliths [12,13]. Severe LIC (lowered fibrinogen level) is common in large VMs of the extremities and rare in cervicofacial VMs [12–16].
A number of events, such as operation, sclerotherapy, prolonged immobilization, menstruation and pregnancy, can trigger the conversion of LIC to disseminated intravascular coagulopathy (DIC), with per- and postoperative bleeding related to consumption of fibrinogen and clotting factors. Therefore, a coagulation profile, with platelet count, fibrinogen and D-dimer levels, should be obtained in any child with VM [9,12,13,17,18].
Elevated D-dimer level associated with a vascular anomaly in an otherwise healthy patient is pathognomonic of a VM [13]. This coagulopathy is completely different from the Kasabach–Merritt phenomenon (KMP) associated with aggressive vascular tumours of infancy (kaposiform haemangio-endothelioma and tufted angioma) [19,20]. Confusion still exists in the literature; many patients with the VM-associated LIC are mislabelled as having ‘haemangioma’ and ‘KMP’, resulting in mismanagement.
Haemarthrosis is particularly troublesome in children with VM-associated chronic LIC, mimicking haemarthrosis seen in haemophilia, and leading to degenerative joint disease and destructive cartilage/bone changes. Symptoms abate with bed rest but, if not treated, this ends in flexion deformity and ankylosis of the joint [9]. These lesions are often mislabelled in the literature as bony, intramuscular or synovial ‘haemangioma’ or ‘cavernous haemangioma’ [21].
Radiological imaging is necessary because a VM is often much more extensive than clinically anticipated. Plain radiography reveals phleboliths as early as the age of 2 years. These round calcifications are pathognomonic of slow venous flow, and caused by intralesional thrombosis (see Fig. 112.6). Radiographs show bony distortion in facial VMs, and bony thinning or periosteal reaction in limb VMs. The latter can mimic osteoid osteoma [21].
Ultrasonography and grey-scale and colour Doppler display the angio-architecture. Most typically, a heterogeneous echogenicity and an ill-defined hypoechoic lacunar pattern are noted; arterial structures are not detected.
Computed tomography scan with iodinated contrast detects soft tissue extension and skeletal alterations. Bony defects are present in about 20% of lesions involving the scalp or forehead.
Magnetic resonance imaging, with spin-echo (SE) T1- and T2-weighted sequences, is the best non-invasive radiological modality to delineate a VM. On SE T2-weighted sequences, VM is made up of well-delineated, often lobulated venous pouches giving a hypersignal. Three-dimensional reconstruction is particularly useful in demonstrating the involved structures. Gadolinium is routinely used for contrast in the MRI study of a VM to differentiate VM from lymphatic malformation (LM) [22]. In cephalic locations, MRI demonstrates VM extending into calvarial bones. The intraosseous venous lakes give an intradiploic hypersignal, sometimes in association with an underlying intracranial dural increased T2 signal. In the case of a knee VM, MRI plus CT provides useful information, and eliminates the need for arthrography before treatment.
Standard and digital computed arteriography visualize VMs poorly.
Phlebography shows a racemose complex of abnormal veins in limb VMs, but not the anatomical location and size of the lesion. Its usefulness is limited in comparison with Doppler and MRI.
Prognosis.
Venous malformations always expand, albeit slowly.
Differential Diagnosis.
A blue VM can be confused with the naevus of Ota or of Ito. Sinus pericranii, with abnormal cerebral venous drainage through a bony cranial defect, from or to the intracranial area, mimics a VM of the forehead or scalp. A dilated vein at the base of the nose is common in newborns, but prominent veins in the frontonasal area are occasionally due to collateral cerebral venous circulation through the cavernous sinus, superior ophthalmic vein, angular vein and facial vein, as a result of a vein of Galen aneurysmal malformation. A deep cephalic VM and an intramuscular limb VM that has atypical MRI findings require a biopsy to rule out sarcoma or neurofibroma. VM deeply located in the neck area should be differentiated from various types of congenital cysts (thyroglossal duct, bronchogenic cyst, etc.). It is important to differentiate between extremity VMs and a complex/combined limb vascular malformation (see below).
Cerebral developmental venous anomaly (DVA) consists of dilated intramedullary veins converging into a large draining vein. This vessel enters either the superficial or the deep system. DVA is an uncommon pathway of cerebral drainage, occurring in 0.05–0.5% of the general population. By contrast, 20% of patients with extensive head and neck VMs have DVA [23], usually consisting of ectatic and dilated veins converging into the drainage system of the deep brain. In contrast with cerebral cavernous malformation, opacification of DVA appears in the venous phase, as do normal veins. CT and MRI with MRA image DVAs. These are usually asymptomatic. Some patients develop headaches, but intracerebral haemorrhage, seizures and neurological deficits do not occur. Therefore, DVA need not be treated.
Treatment.
Selective catheterization of slow-flow vascular lesions is difficult; thus, arterial embolization is seldom used. The best strategy is percutaneous intralesional sclerotherapy [24–29]. Absolute ethanol is the most efficient sclerosing agent. Procedures are performed with real-time fluoroscopic control, and under general anaesthesia with careful monitoring. Local compression or intralesional coil injection is sometimes used to prevent passage into the systemic circulation. Local complications include inflammation, oedema, blistering, necrosis, chronic drainage and scarring, and temporary or permanent nerve deficit. Systemic complications, such as renal or pulmonary toxicity, myocardial depression and even cardiac arrest and death, have been reported [30,31]. Sodium tetradecyl-sulphate or Lauromacrogol 400 injections, given with local anaesthesia or no anaesthesia, are effective for small VMs and cause fewer local side-effects. An acrylic glue is sometimes used preoperatively. Detergent sclerosants as been used as microfoam forms, using air bubbles or carbon dioxide to increase the volume and surface contact with endothelium [32,33]; however, neurological complications have been reported in 2% of patients [34]. More recently, a modified radio-opaque ethanol sclerosing agent has been developed which traps the ethanol within a mesh of ethylcellulose in order to increase its viscosity [35–37]. This decreases the amount of ethanol given and the frequency of possible complications.
For cephalic VM, treatment begins as soon as deformity and functional problems develop. Multiple sessions are required, combining sclerotherapy and surgical procedures, with various timing [38]. Open-bite deformity requires orthodontic management and orthognathic correction after eruption of secondary teeth.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


