As pathognomonic laboratory parameters or histological features do not exist, the diagnosis of infantile SD is established on clinical grounds. In contrast to atopic dermatitis, erythematous patches in SD are clearly demarcated, usually less pruritic, tend to predominate in the flexural folds including the anogenital area.
In patients with systemic symptoms such as diarrhoea or failure to thrive, skin biopsy and further diagnostic steps are warranted to rule out more severe conditions such as immunodeficiencies or metabolic defects [3,4].
Atopic Dermatitis (See Chapter 28)
Despite a period prevalence of 10–20% within the first 6 months of life, atopic dermatitis (AD) does not usually occur in infants younger than 3 months of age and only very rarely causes severe skin symptoms or even erythroderma in neonates [5]. If present, however, acute lesions are characterized by widespread pruritic erythematous papules and patches with marked exudation. Infantile AD typically involves the scalp, the face, the trunk and the extensor surfaces of the extremities whereas, in contrast to infantile psoriasis and seborrhoeic dermatitis, the napkin area is frequently spared [6]. Moreover, in contrast to more serious causes of neonatal erythroderma, newborn infants affected by AD without concurrent food allergy usually thrive and do not exhibit persisting alopecia or additional clinical signs of extracutaneous pathology (e.g. lymphadenopathy, diarrhoea, atypical infections).
The lesions respond rapidly to topical therapy with corticosteroids and emollients. Because of a considerable clinical overlap and rather uncharacteristic histological findings upon skin biopsy, differentiation of erythrodermic AD from other causes of neonatal erythroderma is challenging. Although elevated total serum immunoglobulin E (IgE) levels indicate early onset AD, they are non-specific and may also be associated with more serious disorders such as Omenn syndrome, Wiskott–Aldrich syndrome or Netherton syndrome (see below). Conversely, normal total serum IgE levels do not rule out neonatal or infantile AD because of a high prevalence of the intrinsic variant of AD in this age group [7]. Likewise, recent prospective birth cohort studies have disclosed that, unlike previously postulated, the presence of food-specific IgE is not a reliable indicator of infantile AD, because it can be detected equally often in affected and healthy infants [8]. Hence, if severe AD with concurrent food allergy is suspected in a neonate with erythroderma, the patient should first be fed with an extensively hydrolysed milk- or amino acid-based formula for a limited period of time. Subsequently, the infant should undergo an oral food challenge with the incriminated allergen. Finally, if symptoms recur upon oral food challenge, the diagnosis of food allergy in the context of severe AD is highly probable.
Psoriasis (See Chapter 80)
Up to 16% of children with psoriasis develop symptoms during the first year of life. However, neonatal or congenital psoriasis is very rare. Neonatal erythroderma resulting from psoriasis is likely to evolve into generalized pustular psoriasis (GPP). Erythrodermic GPP requires a thorough diagnostic work-up in order to exclude other serious differential diagnoses, especially infections with herpes simplex virus (HSV), varicella-zoster virus (VZV), Staphylococcus aureus or Candida spp. [9,10]. Moreover, GPP demands immediate treatment to avoid potentially severe complications such as sterile lytic bone lesions, bacterial superinfection or sepsis [11].
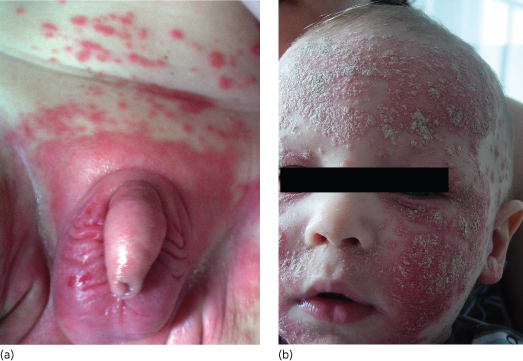
Infantile psoriasis often starts with an unusually severe napkin rash that is better demarcated and brighter red than that of seborrhoeic dermatitis (Fig. 11.2a). Subsequently, rapid dissemination leads to generalized erythematosquamous skin lesions (Fig. 11.2b), but in neonates and young infants psoriatic erythroderma may also mimic non-bullous ichthyosiform erythroderma, severe AD or Netherton syndrome [12]. Hence, correct diagnosis may require skin biopsy that reveals a psoriasiform reaction pattern with laminated parakeratosis, elongated rete ridges often containing neutrophils and little or no dermal lymphocytic infiltrate while LEKTI staining is negative [13]. Once the diagnosis of psoriasis is safely established, topical treatments including use of topical corticosteroid or vitamin D analogues can be used. For more severe or generalized cases, systemic treatment with acitretin (0.5–1 mg/kg/day) has been used with safety and efficacy even in young infants and children with GPP [14].
Fig. 11.2 (a) Non-congenital psoriasis in the napkin area of a male infant revealing sharply demarcated, infiltrated, bright red plaques. (b) Same patient as in (a) showing widespread dissemination of psoriatic lesions with particularly severe erythematosquamous plaques covering the facial and scalp region.
Pityriasis Rubra Pilaris (See Chapter 83)
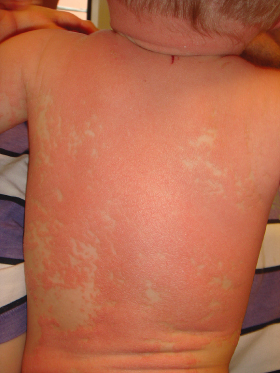
Pityriasis rubra pilaris (PRP) represents a rare papulosquamous disorder of unknown origin that can manifest at any age. In the first month of life, PRP is almost exclusively encountered in those extremely rare patients with congenital disease onset. Clinically resembling atypical juvenile PRP (Griffiths classification type V), skin lesions consist of widespread follicular erythematous papules and patches coalescing into erythroderma. As in typical juvenile PRP, neonates display salmon-coloured erythema with islands of unaffected skin (‘nappes claires’) (Fig. 11.3) whereas palmoplantar keratoderma is usually discrete or completely lacking. Histological features of neonatal PRP have only been reported casuistically and consist of alternating ortho- and parakeratosis with lipping of follicular ostia and associated follicular plugging [15,16].
Fig. 11.3 Atypical juvenile pityriasis rubra pilaris in a male infant with follicular erythematous papules and patches coalescing into erythroderma. Of note, the typically salmon-coloured erythema reveals islands of unaffected skin (‘nappes claires’).
Genodermatoses
Neonatal erythroderma is a presenting sign of several monogenic skin diseases, particularly ectodermal dysplasia and hereditary disorders of keratinization. The latter include isolated ichthyoses with exclusively cutaneous symptoms as well as syndromal ichthyoses associated with potentially severe metabolic, neurological, ophthalmological or other abnormalities [17]. If suspicion arises, skin biopsy is mandatory and further diagnostic steps (e.g. molecular genetics, immunohistochemistry, electron microscopy) should optimally be coordinated in cooperation with national reference centres such as the Network for Ichthyoses and Related Keratinization Disorders: NIRK (http://www.netzwerk-ichthyose.de) or the Centre de Référence National des Maladies Génétiques à Expression Cutanée: MAGEC (http://www.magec.eu).
Isolated Ichthyoses (See Chapter 121)
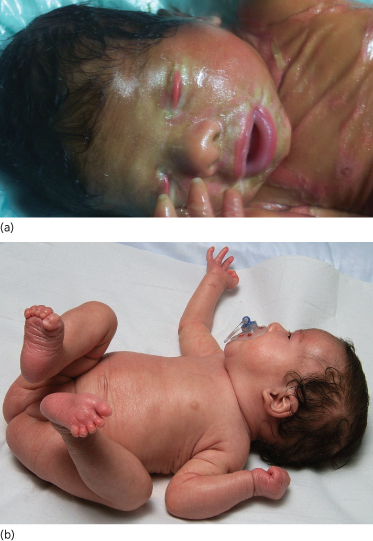
Non-bullous congenital ichthyosiform erythroderma (OMIM 242100) is an autosomal recessive keratinization disorder that is caused by mutations in various genes (TGM1, ALOXE3, ALOX12B, Ichthyin, ABCA12) [18]. As a clinical hallmark, up to 90% of affected patients present with a collodion membrane directly postpartum which is shed during the neonatal period, revealing erythroderma with generalized fine scaling (Fig. 11.4a,b) [19]. While systemic symptoms are usually absent, further cutaneous symptoms can include ectropion or eclabium, palmoplantar keratoses, scarring alopecia or onychodystrophy. At first presentation, it is important to rule out possible differential diagnoses such as Sjögren–Larsson syndrome, Netherton syndrome, trichothiodystrophy, Chanarin–Dorfman syndrome or Gaucher disease type 2 [20].
Fig. 11.4 (a) Collodion baby phenotype in a female neonate with non-bullous congenital erythroderma. From Ott et al. (2008) [1] with permission from Wiley-Blackwell. (b) Same patient as in (a) who reveals mild non-bullous congenital erythroderma with fine scaling and discrete palmoplantar hyperkeratosis after shedding of the collodion membrane.
Autosomal dominant bullous congenital ichthyosiform erythroderma (OMIM 113800), also designated as epidermolytic hyperkeratosis, is caused by mutations in genes encoding the suprabasal keratins 1 and 10 (KRT1, KRT10) [21]. Clinical presentation varies greatly, but affected patients usually develop severe blistering immediately after birth, sometimes with widespread erosions, accompanied by variously severe ichthyosiform erythroderma. During the following weeks, hystrix-like hyperkeratoses appear while marked palmoplantar hyperkeratoses are characteristically encountered in patients with KRT1 mutations only. Initially, other bullous disorders of neonatal onset have to be considered as possible differential diagnoses, especially epidermolysis bullosa hereditaria, ichthyosis bullosa Siemens, staphylococcal scalded skin syndrome and toxic epidermal necrolysis. Affected family members may also facilitate diagnosis, although a negative family history does not rule out bullous congenital ichthyosiform erythroderma because of a rate of >50% of patients with spontaneous mutations that are readily detected in EDTA blood samples or skin biopsy specimens [22].
Syndromal Ichthyoses (See Chapter 121)
Autosomal recessive Chanarin–Dorfman syndrome (OMIM 275630), also referred to as neutral lipid storage disease with ichthyosis, represents a multiorgan disorder elicited by mutations in the ABDH5 gene. These induce pathological alterations in the metabolism of endogenous triglycerides leading to the accumulation of neutral lipids in multiple cell types, particularly leucocytes, myocytes, hepatocytes, fibroblasts and keratinocytes. The resulting clinical phenotype resembles other forms of congenital ichthyosiform erythroderma while systemic manifestations in neonates may consist of subclinical myopathy, sensorineural deafness, cataracts, developmental delay and growth retardation. Lipid vacuoles are present in numerous organs and can be seen in a skin biopsy specimen or in the granulocytes and monocytes of a peripheral blood smear that is visually examined [23,24].
Also known as Conradi–Hünermann–Happle syndrome, type 2 chondrodysplasia punctata (CDPX2, OMIM 302960) is an X-linked dominant disorder caused by mutations in the EBP (emopamil-binding protein) gene. These entail increased serum levels of pathological cholesterol metabolites because of an impaired function of 8-7-sterol isomerase. CDPX2 is nearly always fatal for male fetuses. In contrast, affected female neonates develop severe ichthyosiform erythroderma immediately postpartum which is usually associated with hyperkeratosis and scaling following Blaschko’s lines. Extracutaneous features, which may not become apparent until later during childhood, include frontal bossing, hearing loss, sectorial cataracts, punctate bone calcifications and mild to moderate mental retardation. While elevated serum levels of 8-dehydrocholesterol and a diminished stratum granulosum with cytoplasmic vacuoles upon skin biopsy hint at CDPX2, definitive diagnosis is reached by molecular genetic analysis of the EBP gene [25,26].
The autosomal recessive Netherton syndrome (OMIM 256500) is caused by inactivating mutations in the serine protease inhibitor Kazal-type 5 (SPINK5) gene which codes for the lympho-epithelial Kazal-type 5 related inhibitor (LEKTI). As a result, severe defects in epidermal barrier function and a persistent inflammatory reaction lead to an uncharacteristic neonatal erythroderma while the typical polycyclic serpiginous plaques with erythematous borders and double-edged scaling (ichthyosis linearis circumflexa) usually do not manifest before early childhood. The clinical distinction of Netherton syndrome from other causes of ichthyosiform erythroderma is facilitated by microscopy of hair shafts (scalp hair, eyebrows, eyelashes) revealing trichorrhexis invaginata (‘bamboo hair’) and hypotrichosis. Skin biopsy with immunohistochemical staining reveals a psoriasiform epidermal hyperplasia with absence of the stratum granulosum, a mixed perivascular infiltrate and reduced or absent LEKTI expression. Additionally, total and allergen-specific IgE levels are frequently elevated while other traits of atopic disease such as bronchial asthma, food allergy or allergic rhinitis do not become apparent until late infancy or early childhood. Associated systemic symptoms include hypernatraemic dehydration, failure to thrive and chronic diarrhoea, which are responsible for a fatality rate of up to 20% in the first year of life [13,27,28].
Ectodermal Dysplasia (See Chapter 127)
Erythroderma can also be seen in neonates with ectodermal dysplasias, a heterogeneous group of inherited disorders involving absence or dysplasia of the ectodermal appendages. In particular, neonates with ankyloblepharon–ectodermal dysplasia–clefting syndrome (OMIM 106260), a very rare autosomal dominant genodermatosis caused by TP63 mutations, may present with widespread erythema. In these patients, neonatal erythroderma with extensive erosions and diffuse depigmented patches in previously eroded areas can be observed besides further clinical signs of ectodermal dysplasia such as palmoplantar keratoderma, partial anhidrosis, nail dystrophy, patchy alopecia and hypodontia [29,30].
Other Skin Diseases
Diffuse Cutaneous Mastocytosis (See Chapter 75)
As the rarest subtype of cutaneous mastocytosis, diffuse cutaneous mastocytosis affects less than 2% of patients with cutaneous mastocytosis and usually manifests with a peculiar type of neonatal erythroderma either at birth or shortly thereafter. Characteristically, infiltration of mast cells throughout the entire cutis leads to widespread yellow–reddish erythema accompanied by extensive bullae formation. Other skin manifestations attributable to mediator release from mast cells comprise flushing, urticaria or pachydermia and extracutaneous symptoms may consist of diarrhoea, headache, bone pain or anaphylaxis. Hence, erythrodermic neonates with diffuse cutaneous mastocytosis require interdisciplinary management including bone marrow biopsy if suspicion of systemic mastocytosis arises, as in patients with hepatosplenomegaly, lymphadenopathy, abnormal blood count or clearly elevated serum tryptase levels [31–33].
References
1 El Euch D, Zeglaoui F, Benmously R et al. Erythroderma: a clinical study of 127 cases and review of the literature. Exog Dermatol 2003;2:234–9.
2 Al-Dhalimi MA. Neonatal and infantile erythroderma: a clinical and follow-up study of 42 cases. J Dermatol 2007;34:302–7.
3 Foley P, Zuo Y, Plunkett A, Merlin K, Marks R. The frequency of common skin conditions in preschool-aged children in Australia: seborrhoeic dermatitis and pityriasis capitis (cradle cap). Arch Dermatol 2003;139:318–22.
4 Naldi L, Rebora A. Clinical practice: seborrhoeic dermatitis. N Engl J Med 2009;360:387–96.
5 Wadonda-Kabondo N, Sterne JA, Golding J et al. A prospective study of the prevalence and incidence of atopic dermatitis in children aged 0–42 months. Br J Dermatol 2003;149:1023–8.
6 Eigenmann PA. Clinical features and diagnostic criteria of atopic dermatitis in relation to age. Pediatr Allergy Immunol 2001;12(Suppl 14):69–74.
7 Ott H, Stanzel S, Ocklenburg C et al. Total serum IgE as a parameter to differentiate between intrinsic and extrinsic atopic dermatitis in children. Acta Derm Venereol 2009;89:257–61.
8 Eller E, Kjaer HF, Host A, Andersen KE, Bindslev-Jensen C. Food allergy and food sensitization in early childhood: results from the DARC cohort. Allergy 2009;64:1023–9.
9 Morris A, Rogers M, Fischer G, Williams K. Childhood psoriasis: a clinical review of 1262 cases. Pediatr Dermatol 2001;18:188–98.
10 Lehman JS, Rahil AK. Congenital psoriasis: case report and literature review. Pediatr Dermatol 2008;25:332–8.
11 de Oliveira ST, Maragno L, Arnone M, Fonseca Takahashi MD, Romiti R. Generalized pustular psoriasis in childhood. Pediatr Dermatol 2010;27:349–54.
12 Hoeger PH, Harper JI. Neonatal erythroderma: differential diagnosis and management of the ‘red baby’. Arch Dis Child 1998;79:186–91.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


