Aetiology and Pathogenesis.
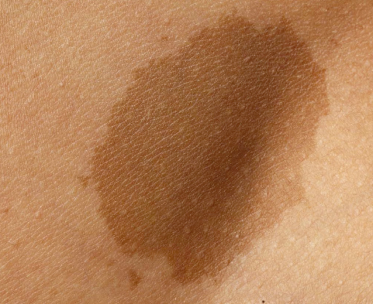
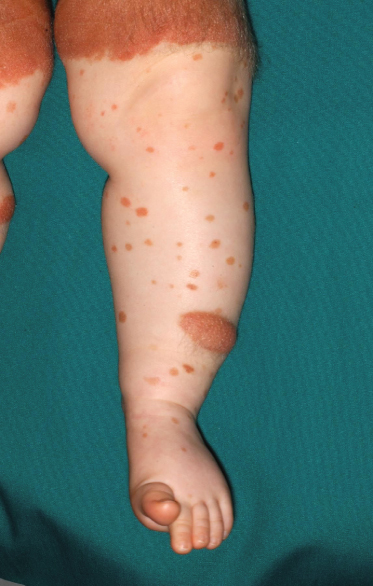
A variety of pigmented lesions of the skin may be obvious at birth. The prevalence varies considerably between series, possibly because of the ethnic mix of the patients examined, as congenital melanocytic naevi may be more common in black or Asian children [2]. These lesions may be difficult to see in the immediate postnatal period, and it may initially be hard to differentiate congenital naevi from café-au-lait spots (Fig. 109.2) clinically. Osburn et al. [3] defined café-au-lait spots as evenly pigmented macular lesions with sharply defined margins and no change in the skin markings. They differentiated naevi by the presence of accentuated skin surface markings, a variation in pigmentation within the lesion, slight palpability of the lesion and variation in sharpness of the margin. The majority of congenital naevi are, however, deeply pigmented at birth.
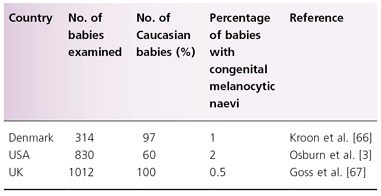
Between 1% and 2% of newborns have congenital melanocytic naevi [3] (Table 109.1). There is very limited sequential information about the eruption of naevi in the period immediately after birth, but it is thought that naevi clinically and histologically similar to the congenital type may erupt within the first year of life. One study suggested that these congenital naevus-like lesions were present in 17% [4] of children and in around 2% of adults [5]. In the only sequential study reported to date of acquired naevi in the same cohort of children [6], 0.5% of 1012 babies examined at birth had melanocytic naevi compared with 35% at the age of 1 year.
Table 109.1 The number of congenital melanocytic naevi detected at birth in three studies
Associated Syndromes – the Carney Complex
There are a variety of hereditary syndromes in which increased numbers of melanocytic naevi occur [7]. The Carney complex is an autosomal dominant (familial) multiple neoplasia syndrome with variable penetrance [8,9] characterized by ‘spotty skin pigmentation’ (lentigines and melanocytic naevi), cardiac and other myxomas, endocrine tumours and melanotic schwannomas [10,11]. In Carney’s original eries, 14/40 cases had cutaneous pigmented lesions, 11/40 had Cushing syndrome associated with multiple adrenocortical nodules with sexual precocity and 29/40 had cardiac myxomas [12]. There are numerous other associated features, such as susceptibility to tumours in sites such as the testis and pituitary. The acronym LAMB, which is also used to describe the signs of the Carney complex, stands for lentigines, atrial myxomas, mucocutaneous myxomas and blue naevi, and Atherton described the same entity as the NAME syndrome in 1980 [13].
The Carney complex is associated with disordered neural crest differentiation and the melanocytic lesions are correspondingly varied: congenital naevi, congenital blue naevi and lentigines (which are commonly centrofacial, on the vermilion border of the lip, or mucosal). Some families map to chromosome 2p, other affected families, have recently been found to have large deletions involving the PRKAR1A gene [14] and it is possible that more families will be found in which the genetic basis of the Carney complex is PRKAR1A.The Carney complex is therefore believed to exist in at least two genetically distinct forms: type 1 linked to chromosome 17 due to mutations in the PRKAR1A gene [9] and type 2 linked to chromosome 2p16 (www.ncbi.nlm.nih.gov).
Congenital melanocytic naevi are said to occur in the epidermal naevus syndrome [12]. There is also a less well-substantiated association with neurofibromatosis [7]. They may also occur in the premature ageing syndromes, in which there is lack of subcutaneous tissue [7].
Pathology.
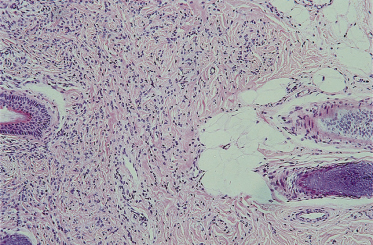
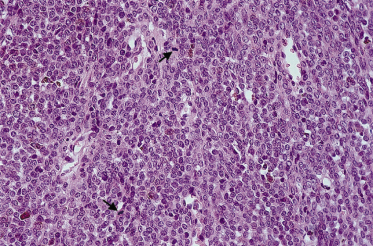
The majority of congenital naevi are dermal or compound melanocytic naevi, with naevus cells typically extending more deeply into the dermis (Fig. 109.3) than those of acquired naevi [15]. The configuration of epidermal melanocytes is similar in congenital and acquired naevi, but congenital naevi more frequently have melanocytes within skin appendages, in smooth muscle, glands, blood vessel walls (Fig. 109.4) and nerves. Melanocytes within striated muscle or fat (see Fig. 109.3) may also be seen [16]. Nickoloff et al. [17] have suggested that the presence of melanocytes within the adventitia of eccrine ducts or hair follicles in the midreticular dermis or deeper is particularly characteristic of congenital melanocytic naevi.
Fig. 109.3 A congenital naevus demonstrating Indian filing of melanocytes within the septae of adipose tissue.
Courtesy of Dr P.H. McKee.
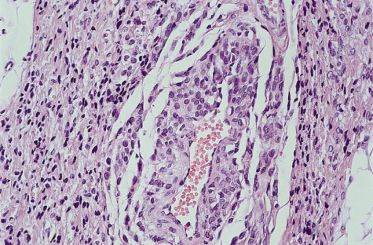
A histological study of naevi biopsied within the first 72 hours of life showed a predominantly junctional localization of melanocytes [18] whereas another study in which naevi were biopsied in older individuals [19] showed a greater degree of deep dermal involvement. Certainly, congenital naevi appear to mature from flat junctional naevi to palpable dermal naevi (see Fig. 109.1) in time. It was therefore suggested that congenital naevi evolve within the first few months of life, from junctional to deep lesions. It was thought that this transition offered the possibility of effective early treatment with dermabrasion [20]. However, Nickoloff’s subsequent study [17] showed that deep melanocytes are demonstrable in the majority of congenital naevi within 72 hours of birth. The authors suggested that differences in the histological appearances of congenital naevi in different studies represented in utero variation rather than any change with time after birth.
There are a variety of proliferative neoplasms that may develop in congenital melanocytic naevi, which cause great consternation and confusion. Figure 109.5 shows a deep dermal mitosis in a congenital lesion. Certainly, malignant melanoma may arise but can be difficult to distinguish from other proliferative neoplasms. These can look malignant but seem to behave in a more benign manner [21]. In addition, a rapidly growing, ulcerative tumour called nodular proliferative neurocristic hamartoma has been described at birth [22] or later in life [23]. This fetal hamartoma consists of diverse tissues of neuroectodermal and ectomesenchymal origin. Hamartomatous proliferations of various types may also develop [24]. The greatest diagnostic difficulty occurs with lesions that simulate melanoma (characterized by the proliferation of epithelioid melanocytes), so that attempts have been made to categorize these variants. The demonstration of aneuploidy in these lesions [25,26] is suggestive of a malignant potential, although Mancianti et al. [22] failed to demonstrate a malignant phenotype in two cases in vitro. A recent genotypic study suggested that large congenital naevi commonly have NRAS mutations (19 out of 27) whereas BRAF mutations were less common (4/27 15%) [27]. This is in contrast with acquired melanocytic naevi which tend to have BRAF mutations more commonly than NRAS mutations [28].
Fig. 109.5 Two mitoses are visible (marked with arrows), deep within a congenital naevus.
Courtesy of Dr P.H. McKee.
Clinical Features.
Congenital naevi at birth are usually pigmented, although they vary in colour from light brown to a slate black .The colours within may vary, but there is usually little erythema. In some texts, it is stated that congenital naevi may darken with time but, in the author’s experience, within the first year the majority become significantly paler at all sites (Fig. 109.6), especially on the scalp [29]. Very rarely, there may be significant, spontaneous, clinical disappearance of a naevus on the scalp [29] (Fig. 109.7), although there may be histological persistence of the naevus [30]. The majority of the naevi are palpable but reasonably flat at birth. With time, the naevi may become more palpable and often become rugose or mammillated, with adverse effects on the cosmetic appearance. This is analogous to the process of maturation seen with time in acquired naevi, which commonly develop papillomas or nodules, which may be pale in colour. Less frequently, the congenital naevus may develop large overgrowths simulating plexiform neurofibromas. With time, many congenital naevi develop terminal hair, which may also be deeply pigmented (Fig. 109.8). These changes with time are variable and difficult to predict. A significant proportion remain macular.
Fig. 109.6 A giant congenital pigmented naevus on the trunk showing (a) deep pigmentation at birth that faded to leave a paler naevus (b) yet showing thickening and growth of terminal hair.
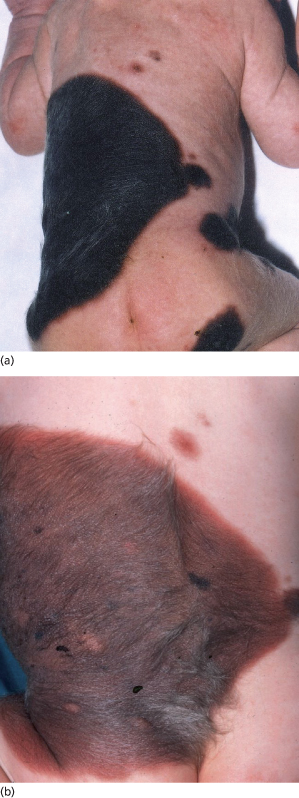
Fig. 109.7 A congenital naevus of the scalp, which was camouflaged by hair and then faded away in the first few years of life.

Fig. 109.8 A small congenital naevus. Such naevi are commonly much larger than acquired naevi, raised and deeply pigmented.
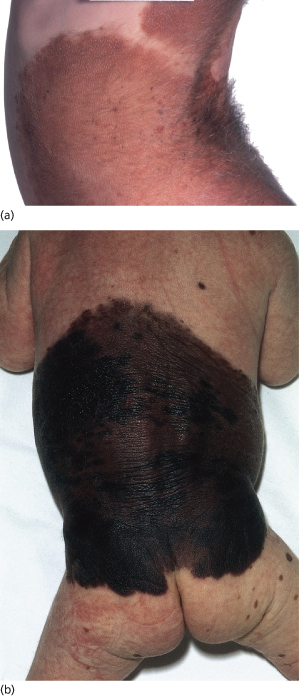
Congenital naevi tend to be much larger in diameter than acquired naevi. Most congenital naevi are at least 5 mm in diameter (see Fig. 109.4) even at birth, whereas the majority of acquired naevi never grow to more than 5 mm in diameter. Most of the melanomas that evolve from naevi occur in large naevi and therefore a distinction has been drawn between congenital naevi less than 10 cm in diameter and those over 10 cm. Most authors use the American National Institutes of Health (NIH) consensus definition to categorize these naevi according to size. In this document, small naevi are defined as under 1.5 cm in diameter, large naevi as having a diameter between 1.5 and 20 cm and giant naevi as having a diameter of 20 cm or more [31]. Giant naevi, covering large parts of the body are fortunately rare. Castilla et al. [2] reported a study of 500,000 infants in South America, in which one naevus at least 10 cm in diameter was identified per 20,445 subjects. Such naevi are often known as giant, garment or bathing trunk naevi (Fig. 109.9, see Fig. 109.6). A rarer variant is the giant cerebriform melanocytic naevus [32], which is usually seen on the scalp. This type is associated with excessive growth of furrowed skin so that the differential diagnosis on the scalp is that of cutis verticis gyrata.
Fig. 109.9 (a) A mature giant congenital naevus that faded over time. Whilst still very obvious and bearing terminal hair, the skin is elastic and the child well adapted to it. (b) A child with a giant melanocytic naevus.
Courtesy of Professor John Harper.
Dermoscopy of congenital naevi has been studied and some authors have described reticular, globular or reticuloglobular forms [33]. Others have suggested that congenital naevi have some particular characteristics: ‘focal thickening of network lines, globules, target globules, homogeneous diffuse pigmentation, hyperpigmented areas, blotches and target vessels’ [34]. The patterns that would cause concern, however, are similar to acquired naevi, namely the presence of irregular blue/white colours, irregularity of the pigment network, and dots, globules and red/brown structures indicative of vascularization.
Complications of Large Congenital Naevi.
One of the most important complications of naevi is melanoma, which is discussed below. So-called satellite naevi develop over time in many children with giant naevi: multiple small congenital type naevi appear at many body sites. Some are obvious at birth and some develop in the first few years (tardive naevi) (Fig. 109.10). Satellite naevi were reported in 74% of one series of children with giant naevi [35] and in a second series, 83% had such naevi at birth [36]. In the Ruiz-Maldonado series, 31% of the patients also had naevi in mucous membranes [35]. The absence of satellite naevi was a good prognostic factor for a lack of complications in a recent series of children with congenital naevi from the Great Ormond Street Hospital, London, UK [36].
Patients may have great concerns about aesthetic effects if the naevi are large and/or on exposed skin, but it is the author’s experience that many children make a very good psychological adjustment to their appearance if very well supported. In this regard, there are online support networks available for families [37]: www.nevussupport.com, www.nevusnetwork.org, www.birthmarksupportgroup.org.uk.
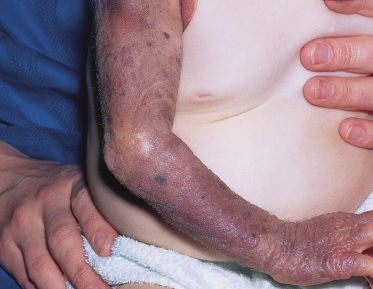
Commonly, there are reduced subcutaneous tissues associated with the naevus, which may be most symptomatic over the sacrum. The naevus may also disrupt normal structures within the skin so that the epidermis can be lifted off the dermis by relatively mild trauma. Affected limbs are commonly smaller in girth but not in length and are usually functionally normal [35,38] (Fig. 109.11).
Naevi overlying the head, neck or spine may be accompanied by neurocutaneous melanosis, in which naevus cells occur within the central nervous system and meninges [39,40], resulting in raised intracranial pressure, hydrocephalus or space-occupying spinal lesions. Rare cases are reported with gross hydrocephalus [41] which may cause the Dandy–Walker syndrome [42], although in a series of 80 Mexican patients with giant naevi, only one case of hydrocephalus was reported [35]. Although symptomatic neuromelanosis is rare, abnormalities on electro-encephalogram or magnetic resonance imaging (MRI) are more common. In the Great Ormond Street Hospital series [36], 15% of 120 albeit selected patients had abnormal findings at MRI. Of these, 15/18 had melanosis, 3/18 had benign tumours, 2/18 had hydrocephalus and 1/18 had a central nervous system malformation; 13/18 of the patients were symptomatic by the age of 2 years but the others remained asymptomatic at follow-up. Seven of 18 ultimately required neurosurgical intervention. One of the children died; he had a small amount of melanosis initially but developed progressive neurological symptoms at the age of 15 months, which occurred as a result of continued proliferation of the melanocytes within the central nervous system.
In a recent UK series, abnormal neurodevelopment was reported in 19% of 349 patients with congenital naevi of a range of sizes (43% <10 cm in diameter, others being larger) [43]. Speech delay was the most common problem reported and 4% had fits. These data are difficult to assess in what is likely to be a selected series. Foster and colleagues have reported that symptomatic neuromelanosis is rare [44].
Melanoma Arising in Congenital Naevi
Melanoma is the most significant complication of congenital naevi but the degree of risk remains controversial, largely because most of the reported series come from large referral centres and may therefore suffer from bias of ascertainment.
There seems to be no doubt that there is an increased risk of melanoma in patients with very large naevi. In the series by Swerdlow et al. [45], two melanomas, both fatal, were reported in 265 patients with naevi covering at least 5% of their body area. One melanoma occurred at the age of 18 years and the other at the age of 20 years. In a second series of 80 patients, four melanomas occurred [35] (two in children under the age of 3 years), three of which were fatal. In the most recent series only one fatal melanoma occurred in 224 patients studied with a mean follow-up of 8 years [36], although 5/349 patients reported in a second paper from the same institution with overlapping cases (mean prospective follow-up of 9.2 years) developed melanoma (1.4%) [43].
Therefore, in giant naevi there appears to be a definite (but probably slight) risk and the age of onset of the melanoma is considerably earlier than in the general population. In contrast with the norm, tumours may arise from below the dermoepidermal junction in congenital naevi, which may delay diagnosis [46]. There have been several attempts to estimate lifetime risk, giving values between 4.6% and 14% [47]. Krengel et al. performed a useful review of a total of 6571 patients with congenital melanocytic naevi, in a number of studies with variable follow-up (the range of mean periods of follow-up was 3.4–23.7 years). Overall, 46 patients (0.7%) developed a total of 49 melanomas [48]. The mean age at diagnosis of melanoma was 15.5 years (median 7 years). Primary melanomas arose inside the naevi in 33 of 49 cases (67%). In seven cases (14%), metastatic melanoma with an unknown primary was encountered; in four cases (8%) the melanoma developed at an extracutaneous site. The risk of developing melanoma and the fatality rate were highest by far in congenital melanocytic naevi 40 cm or greater in diameter. A much larger series, or a meta-analysis of several series, would be necessary to produce a realistic quantification of lifetime risk.
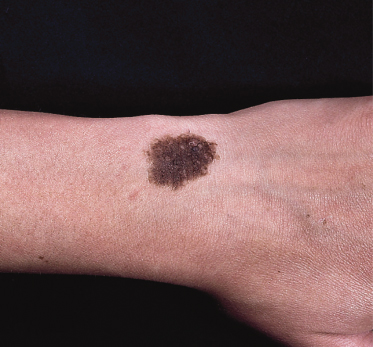
There are numerous case reports of melanoma arising in smaller congenital naevi [49] (Fig. 109.12), but such naevi are relatively common and it is difficult from the studies reported so far to compare the relative risk of melanoma arising from a small congenital naevus with that of a melanoma arising in an acquired naevus. Various estimates have been made. Rhodes and co-workers [49,50] estimated a lifetime risk of between 0.8% and 4.9% in naevi of less than 4.5 cm in diameter, depending on whether histological or reported history was used to define a ‘congenital’ naevus. Swerdlow et al. [45] failed to demonstrate any increased risk for naevi covering less than 5% of the body. It seems unlikely that the true risk from a small congenital naevus will ever be established accurately because of the impossibility of gathering a sufficiently large cohort of patients whose congenital naevi have been left in situ.
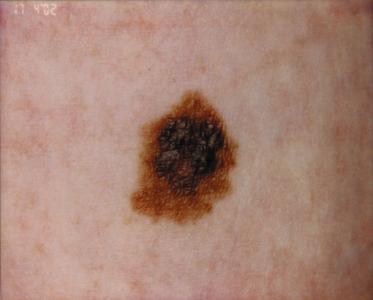
Rarely, intracerebral primary melanoma may complicate leptomeningeal melanocytosis [51].
Treatment.
A magnetic resonance imaging (MRI) scan should be considered in babies with naevi over the cranium or spine, or extensive naevi generally [36], to exclude significant leptomeningeal melanocytosis. However, although MRI anomalies are common, occurring in 15% [36] to 30% [44] of selected series, symptomatic neurocutaneous melanosis is rare [44]. The value of scanning therefore seems questionable in the absence of neurological signs, although it is often recommended. Regular neurological examination, however, is clearly important.
The aims of treatment are to improve the cosmetic effect of naevi and to reduce the risk of malignant transformation. To date, there is no proof that any treatment regime fulfils the second aim for giant naevi in which there is no possibility of complete excision because of the surface area of skin involved and the depth of the naevus within the skin and subcutis. There is perhaps a stronger argument for prophylactic excision of large naevi where complete excision of the naevus is technically possible with a reasonable cosmetic result.
The potential benefits for the child in terms of cosmesis must be balanced with short- and long-term risks. Small naevi are not, in the UK at least, routinely excised prophylactically [52] because the risk of malignant change is not established. Parents should, however, be advised to keep the naevus under review and to seek advice if the naevus changes in shape, size or colour. As such naevi can change through life in an entirely benign way, education about the nature of change that would cause concern is essential. Photographs of the child’s naevus and images of melanomas are useful for the family to take home. It would seem reasonable to excise small naevi prophylactically, when they are difficult to keep under review, such as those on the scalp or in the middle of the back, particularly if they have any atypical features such as intense or irregular pigmentation that persists beyond infancy.
The most common treatment for larger naevi is surgery using a variety of techniques such as multistep surgery, tissue expanders and grafting (see Chapter 191). The choice of surgical technique depends on the site of the naevus. A review by Bauer & Vicari [53] summarizes the preferred surgical approaches taken at the Children’s Hospital in Chicago in 1988, which are similar to those taken currently in the UK, i.e. tissue expansion is used for the head and neck, but at other sites excision and grafting are more usual, with consequently poorer cosmetic results. No statistics are available regarding the proportion of children who are treated by surgery but in the author’s experience in the UK the majority are not treated.
The cosmetic results of surgery for giant naevi on the trunk are generally poor, and there have been attempts to improve results, mainly by treatment within the first few weeks of life by dermabrasion or curettage [54]. The rationale for this approach was originally the hypothesis that melanocytes are more superficial at birth [20]. Also, more recently, it has become accepted that there may be a natural plane of cleavage within the upper dermis very early in life, which can be exploited by the curette to reduce the risk of subsequent scarring [55]. It is difficult from the literature to assess the long-term cosmetic results of the approach of curettage, but the results of dermabrasion seem to be disappointing. A review of 215 patients treated in Germany reported hypertrophic scars in 15% (particularly on the lower back) and good permanent reduction in pigmentation in only 34%, which was more likely when treatment was carried out early [56]. Among a smaller series of 12 neonates treated by dermabrasion, good results were reported in 10, although six went on to have surgery later [57].
Other ablative techniques have been tried, such as cryotherapy and more recently the Q-switched ruby laser. This laser emits light at 694 nm, which is relatively selectively taken up by melanin. There is a report of valuable cosmesis [58] with the use of this laser (see Chapter 191). There is no evidence that the use of visible light to treat such naevi is associated with any risk in terms of carcinogenesis [59] but concerns have been expressed that there is a potential risk [60].
Overall, then, decisions about intervention in children born with large or giant naevi are made on the basis of balancing the risk of developing melanoma (which is poorly quantified), the cosmetic deficit of the naevus and the risks of surgery. Furthermore, the long-term cosmetic results of surgery are often disappointing, although children in one study reported a preference for a surgical scar over their naevus [61]. Where surgery is avoided, the skin remains elastic and functional, and in a good proportion the colour fades over time (see Fig. 109.9b) so that the author’s advice is usually to avoid surgery. However, the psychological impact of giant congenital naevi can be great and a risk of melanoma exists, so there is a need to establish better approaches to removal. Those available currently leave much to be desired. The decision as to whether to operate or not may be very difficult for families. Access to before-and-after photographs from previous cases may help in this process.
Genetic Counselling
Congenital naevi are essentially considered to be developmental abnormalities of the skin, with a very low risk of recurrence in subsequent pregnancies, which is an important source of reassurance for parents who have a child with a giant melanocytic naevus [62]. A large congenital naevus was reported in four second-degree relatives of 80 patients with giant naevi in a study by Ruiz-Maldonado et al. [35]. Moreover, small congenital naevi appear to aggregate in some families [63] and in a recent series, 9% of second-degree relatives of patients with congenital naevi were said by the families to have also had congenital naevi [43]. The possibility therefore remains that inherited genes may play a role, although it seems most likely that giant congenital naevi arise as a result of a lethal mutation surviving only by mosaicism [64] and that paradominant inheritance might explain rare clustering in families [65], i.e. heterozygous individuals would be unaffected and a congenital naevus would become clinically obvious only as a result of a postzygotic mutation in utero of the previously wild type, or healthy gene, to create a mosaic (see Chapter 115).
References
1 Linabery AM, Ross JA. Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975–1999. Cancer 2008;113:2575–96.
2 Castilla EE, da Graca Dutra M, Orioli–Parreiras IM. Epidemiology of congenital pigmented naevi: I. Incidence rates and relative frequencies. Br J Dermatol 1981;104:307–15.
3 Osburn K, Schosser RH, Everett MA. Congenital pigmented and vascular lesions in newborn infants. J Am Acad Dermatol 1987;16:788–92.
4 Gallus S, Naldi L. Distribution of congenital melanocytic naevi and congenital naevus-like naevi in a survey of 3406 Italian schoolchildren. Br J Dermatol 2008;159:433–8.
5 Kopf AW, Levine LJ, Rigel DS et al. Prevalence of congenital-nevus-like nevi, nevi spili, and cafe au lait spots. Arch Dermatol 1985;121:766–9.
6 Goss B, Forman D, Ansell P et al. The prevalence and characteristics of congenital pigmented lesions in newborn babies in Oxford. Paed Perinatal Epidemiol 1990;4:448–57.
7 Marghoob AA, Orlow SJ, Kopf AW. Syndromes associated with melanocytic nevi. J Am Acad Dermatol 1993;29:373–88;quiz 88–90.
8 Sasaki A, Horikawa Y, Suwa T et al. Case report of familial Carney complex due to novel frameshift mutation c.597del C (p.Phe200LeufsX6) in PRKAR1A. Mol Genet Metab 2008;95:182–7.
9 Mateus C, Palangie A, Franck N et al. Heterogeneity of skin manifestations in patients with Carney complex. J Am Acad Dermatol 2008;59:801–10.
10 McKusick V. Online Mendelian Inheritance in Man. 2002. www.ncbi.nlm.nih.gov/omim
11 Kirschner LS, Sandrini F, Monbo J et al. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum Mol Genet 2000;9:3037–46.
12 Carney JA, Gordon H, Carpenter PC et al. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985;64:270–83.
13 Atherton DJ, Pitcher DW, Wells RS et al. A syndrome of various cutaneous pigmented lesions, myxoid neurofibromata and atrial myxoma: the NAME syndrome. Br J Dermatol 1980;103:421–9.
14 Horvath A, Bossis I, Giatzakis C et al. Large deletions of the PRKAR1A gene in Carney complex. Clin Cancer Res 2008;14:388–95.
15 Rhodes AR, Silverman RA, Harrist TJ et al. A histologic comparison of congenital and acquired nevomelanocytic nevi. Arch Dermatol 1985;121:1266–73.
16 Lever W, Schaumburg-Lever G. Melanocytic nevus. In: Histopathology of the Skin. Philadelphia: J B Lippincott, 1990: 756–805.
17 Nickoloff BJ, Walton R, Pregerson-Rodan K et al. Immunohistologic patterns of congenital nevocellular nevi. Arch Dermatol 1986;122:1263–8.
18 Walton RG, Jacobs AH, Cox AJ. Pigmented lesions in newborn infants. Br J Dermatol 1976;95:389–96.
19 Mark GJ, Mihm MC, Liteplo MG et al. Congenital melanocytic nevi of the small and garment type. Clinical, histologic, and ultrastructural studies. Hum Pathol 1973;4:395–418.
20 Johnson H. Permanent removal of pigmentation from giant hairy naevi by dermabrasion in early life. Br J Plast Surg 1977;30:321–3.
21 Borges AF, Lineberger AS. Malignant melanoma without metastasis in a giant nevus. Ann Plast Surg 1984;12:454–60.
22 Mancianti ML, Clark WH, Hayes FA et al. Malignant melanoma simulants arising in congenital melanocytic nevi do not show experimental evidence for a malignant phenotype. Am J Pathol 1990;136:817–29.
23 Mezebish D, Smith K, Williams J et al. Neurocristic cutaneous hamartoma:a distinctive dermal melanocytosis with an unknown malignant potential. Mod Pathol 1998;11:573–8.
24 Drut R. Polypoid dermal dendrocytic hamartoma in a giant congenital melanocytic nevus. Int J Surg Pathol 2007;15:73–6.
25 Stenzinger W, Suter L, Schumann J. DNA aneuploidy in congenital melanocytic nevi: suggestive evidence for premalignant changes. J Invest Dermatol 1984;82:569–72.
26 Newton JA, Camplejohn RS, McGibbon DH. A flow cytometric study of the significance of DNA aneuploidy in cutaneous lesions. Br J Dermatol 1987;117:169–74.
27 Dessars B, de Raeve LE, Morandini R et al. Genotypic and gene expression studies in congenital melanocytic nevi: insight into initial steps of melanotumorigenesis. J Invest Dermatol 2009;129:139–47.
28 Pollock PM, Harper UL, Hansen KS et al. High frequency of BRAF mutations in nevi. Nat Genet 2003;33:19–20.
29 Strauss RM, Newton Bishop JA. Spontaneous involution of congenital melanocytic nevi of the scalp. J Am Acad Dermatol 2008;58:508–11.
30 Vilarrasa E, Baselga E, Rincon C et al. Histologic persistence of a congenital melanocytic nevus of the scalp despite clinical involution. J Am Acad Dermatol 2008;59:1091–2.
31 Conference C. Precursors to malignant melanoma. JAMA 1984;251:1864–6.
32 Quaedvlieg PJ, Frank J, Vermeulen AH et al. Giant ceribriform intradermal nevus on the back of a newborn. Pediatr Dermatol 2008;25:43–6.
33 Changchien L, Dusza SW, Agero AL et al. Age- and site-specific variation in the dermoscopic patterns of congenital melanocytic nevi: an aid to accurate classification and assessment of melanocytic nevi. Arch Dermatol 2007;143:1007–14.
34 Ingordo V, Iannazzone SS, Cusano F et al. Dermoscopic features of congenital melanocytic nevus and Becker nevus in an adult male population: an analysis with a 10-fold magnification. Dermatology 2006;212:354–60.
35 Ruiz-Maldonado R, Tamayo L, Laterza AM et al. Giant pigmented nevi: clinical, histopathologic, and therapeutic considerations. J Pediatr 1992;120:906–11.
36 Kinsler VA, Chong WK, Aylett SE et al. Complications of congenital melanocytic naevi in children: analysis of 16 years’ experience and clinical practice. Br J Dermatol 2008;159:907–14.
37 Krengel S, Breuninger H, Hauschild A et al. Installation of a network for patients with congenital melanocytic nevi in German-speaking countries. J Dtsch Dermatol Ges 2008;6:204–8.
38 Itin PH, Lautenschlager S. Lower and upper extremity atrophy associated with a giant congenital melanocytic nevus. Pediatr Dermatol 1998;15:287–9.
39 Slaughter JC, Hardman JM, Kempe LG et al. Neurocutaneous melanosis and leptomeningeal melanomatosis in children. Arch Pathol 1969;88:298–304.
40 Marghoob AA, Schoenbach SP, Kopf AW et al. Large congenital melanocytic nevi and the risk for the development of malignant melanoma. A prospective study. Arch Dermatol 1996;132:170–5.
41 Amer A, Fischer H. Giant congenital melanocytic nevi. Clin Pediatr (Phila) 2008;47:824–6.
42 Schreml S, Gruendobler B, Schreml J et al. Neurocutaneous melanosis in association with Dandy–Walker malformation: case report and literature review. Clin Exp Dermatol 2008;33:611–14.
43 Kinsler VA, Birley J, Atherton DJ. Great Ormond Street Hospital for Children Registry for Congenital Melanocytic Naevi: prospective study 1988–2007. Part 1 epidemiology, phenotype and outcomes. Br J Dermatol 2009;160(1):143–50.
44 Foster RD, Williams ML, Barkovich AJ et al. Giant congenital melanocytic nevi: the significance of neurocutaneous melanosis in neurologically asymptomatic children. Plast Reconstr Surg 2001;107:933–41.
45 Swerdlow AJ, English JS, Qiao Z. The risk of melanoma in patients with congenital nevi: a cohort study. J Am Acad Dermatol 1995;32:595–9.
46 Reed RJ, Martin P. Variants of melanoma. Semin Cutan Med Surg 1997;16:137–58.
47 Illig L, Weidner F, Hundeiker M et al. Congenital nevi less than or equal to 10 cm as precursors to melanoma. 52 cases, a review, and a new conception. Arch Dermatol 1985;121:1274–81.
48 Krengel S, Hauschild A, Schafer T. Melanoma risk in congenital melanocytic naevi: a systematic review. Br J Dermatol 2006;155:1–8.
49 Rhodes AR, Sober AJ, Day CL et al. The malignant potential of small congenital nevocellular nevi. An estimate of association based on a histologic study of 234 primary cutaneous melanomas. J Am Acad Dermatol 1982;6:230–41.
50 Rhodes AR, Melski JW. Small congenital nevocellular nevi and the risk of cutaneous melanoma. J Pediatr 1982;100:219–24.
51 Slaughter J, Hardman J, Kemple L et al. Neurocutaneous melanosis and leptomeningeal melanocytosis in children. Arch Dermatol 1969:298–304.
52 Newton Bishop JA, Corrie PG, Evans J et al. UK guidelines for the management of cutaneous melanoma. Br J Plast Surg 2002;55:46–54.
53 Bauer BS, Vicari FA. An approach to excision of congenital giant pigmented nevi in infancy and early childhood. Plast Reconstr Surg 1988;82:1012–21.
54 Miller CJ, Becker DW Jr. Removing pigmentation by dermabrading naevi in infancy. Br J Plast Surg 1979;32:124–6.
55 De Raeve LE, de Coninck AL, Dierickx PR et al. Neonatal curettage of giant congenital melanocytic nevi. Arch Dermatol 1996;132:20–2.
56 Rompel R, Moser M, Petres J. Dermabrasion of congenital nevocellular nevi: experience in 215 patients. Dermatology 1997;194:261–7.
57 Bohn J, Svensson H, Aberg M. Dermabrasion of large congenital melanocytic naevi in neonates. Scand J Plast Reconstr Surg Hand Surg 2000;34:321–6.
58 Goldberg DJ, Stampien T. Q-switched ruby laser treatment of congenital nevi. Arch Dermatol 1995;131:621–3.
59 Chan HH, Yang CH, Leung JC et al. An animal study of the effects on p16 and PCNA expression of repeated treatment with high-energy laser and intense pulsed light exposure. Lasers Surg Med 2007;39:8–13.
60 Mahmoud BH, Hexsel CL, Hamzavi IH et al. Effects of visible light on the skin. Photochem Photobiol 2008;84:450–62.
61 Koot HM, de Waard-van der Spek F, Peer CD et al. Psychosocial sequelae in 29 children with giant congenital melanocytic naevi. Clin Exp Dermatol 2000;25:589–93.
62 Goodman RM, Caren J, Ziprkowski M et al. Genetic considerations in giant pigmented hairy naevus. Br J Dermatol 1971;85:150–7.
63 Rhodes AR, Slifman NR, Korf BR. Familial aggregation of small congenital nevomelanocytic nevi. Am J Med Genet 1985;22:315–26.
64 Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol 1987;16:899–906.
65 Danarti R, Konig A, Happle R. Large congenital melanocytic nevi may reflect paradominant inheritance implying allelic loss. Eur J Dermatol 2003;13:430–2.
66 Kroon S, Clemmensen OJ, Hastrup N. Incidence of congenital melanocytic nevi in newborn babies in Denmark. J Am Acad Dermatol 1987;17:422–6.
67 Goss BD, Forman D, Ansell PE et al. The prevalence and characteristics of congenital pigmented lesions in newborn babies in Oxford. Paediatr Perinat Epidemiol 1990;4:448–57.
Café-au-lait Macules
Definition.
Café-au-lait macules are impalpable, lightly pigmented cutaneous lesions (see Fig. 109.2) in which, histologically, there is an increase in basal cell melanin and there may be giant melanosomes. There is no proliferation of basal melanocytes.
Incidence.
Less than 2% of babies have one or more café-au-lait macules [1,2]. They are much more common (28%) in children aged between 6 and 18 years [3], and in adults (14% in one series [4]).
Clinical Features.
These lesions are pale and evenly pigmented and may have a rounded or angulated shape. Café-au-lait macules are common and, in the vast majority of cases, the macules are few in number with no general health implications. Where they occur in larger numbers, their presence may be indicative of a variety of congenital or hereditary syndromes.
The most common underlying condition is neurofibromatosis type 1 (NF1) so children with multiple lesions should be examined for the presence of axillary freckling and Lisch nodules (pigmented hamartomas of the iris), which would confirm the diagnosis of NF1. Lisch nodules are present in 80% of NF1 patients of all ages (see Chapter 128) [5].
However, a number of other traits are also associated with multiple café-au-lait spots. Several families with multiple café-au-lait macules and no other evidence of NF1 have been described. The association of café-au-lait macules with pulmonary stenosis and learning difficulties was reported by Watson [6]. Watson syndrome is now mapped to chromosome 17 and is thought probably to be allelic with NF1.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


