History.
The earliest well-documented case of porphyria probably is that of King George III of England, who had periodic attacks of abdominal pain, tachycardia, discoloured urine and ‘madness’; he was permanently affected by this condition from 1811 until his death in 1820 [1]. In 1874, a patient with photosensitivity, splenomegaly and dark urine was described by Schultz. In 1911, Günther was the first to suggest that the dark urine in these patients was caused by the presence of porphyrin; 4 years later, Fischer identified the porphyrin to be a natural porphyrin, which he named uroporphyrin as it was obtained from the urine. Between 1921 and 1979, all the currently known types of porphyrias were described (Table 107.1) [2].
Table 107.1 History of the porphyrias
Aetiology
Haem Biosynthetic Pathway and Enzyme Deficiencies in Porphyrias
Liver and bone marrow are the two major sites of haem biosynthesis. Specific deficiency of the enzymes of haem biosynthetic pathways has been identified in hepatic or erythropoietic tissue, or in both, in all types of porphyrias. The pathway and the defective enzymes associated with the porphyrias are discussed below (see Fig. 107.1). cDNA for all the enzymes in the haem biosynthetic pathway has now been cloned, and chromosomal locations of all the genes have been assigned (Table 107.2) [3,4].
Table 107.2 Chromosomal location of genes of haem biosynthetic pathway enzymes*
| Porphyria | Enzyme | Chromosomal location |
| – | 5-Aminolaevulinic acid synthase (ALAS) | 3p21.1 (housekeeping gene) Xp11.21 (erythroid-specific gene) |
| ALA dehydratase deficiency porphyria (ADP) | 5-Aminolaevulinic acid (ALAD) | 9p34 |
| Acute intermittent porphyria (AIP) | Porphobilinogen deaminase (PBGD) | 11q23.3 |
| Congenital erythropoietic porphyria | Uroporphyrinogen III synthase (UROS) | 10q25.2 r → q26.3 |
| Porphyria cutanea tarda (PCT)/hepatoerythropoietic porphyria (HEP) | Uroporphyrinogen decarboxylase (UROD) | 1p34 |
| Hereditary coproporphyria (HCP) | Coproporphyrinogen oxidase (CPO) | 3q12 |
| Porphyria variegata (PV) | Protoporphyrinogen oxidase (PPO) | 1q23 |
| Erythropoietic protoporphyria (EPP) | Ferrochelatase | 18q21.3 |
*Modified from Sassa 2002 [4] and Riddle et al. 1989 [5].
All porphyrinogens formed in the pathway can undergo spontaneous oxidation to their corresponding porphyrins, detectable in biological specimens (Fig. 107.2). Although oxidation of protoporphyrinogen IX to protoporphyrin can occur spontaneously, it can also be mediated by protoporphyrinogen oxidase. All porphyrins are phototoxic compounds, accounting for the cutaneous manifestations of porphyrias. The number of carboxyl side-chains in the porphyrin molecule determines the physical properties of these molecules, with uroporphyrin, an 8-carboxyl porphyrin, being hydrophilic, and protoporphyrin, a 2-carboxyl porphyrin, being lipophilic. The commonly used synonyms for porphyrins are shown in Box 107.2.
Box 107.2 Commonly Used Synonyms
| • Uroporphyrin | 8-Carboxyl porphyrin |
| • Coproporphyrin | 4-Carboxyl porphyrin |
| • Harderoporphyrin | 3-Carboxyl porphyrin |
| • Protoporphyrin | 2-Carboxyl porphyrin |
5-Aminolaevulinate Synthase
This first enzyme of the haem biosynthetic pathway, 5-aminolaevulinate synthase (ALAS), condenses glycine and succinyl co-enzyme A (CoA) to form aminoketone ALA, requiring pyridoxal 5′-phosphate as a co-factor. This enzyme is located at the mitochondrial membrane. There are non-erythroid (ALAS1) and erythroid (ALAS2) isoforms, encoded by two separate genes [5]. The housekeeping gene encoding ALAS1 is located on chromosome 3p21.1, and the erythroid-specific gene encoding ALAS2 is located on chromosome Xp11.21 (see Table 107.2) [3,4,6]. Both genes have 11 exons, the former encoding for a protein of 640 amino acid residues and the latter 587 amino acid residues. ALAS is an inducible enzyme.
Clinical Significance
ALA synthase is an inducible enzyme that is present at the lowest concentration among all enzymes in the haem biosynthetic pathway. It is therefore intimately involved in the regulation of haem biosynthesis. The activity of hepatic ALAS1 is increased in the liver of patients with acute porphyrias and mixed porphyrias upon exposure to agents that precipitate visceral and neurological attacks. Furthermore, synthesis of hepatic ALAS1 is modulated by feedback inhibition by haem; in contrast, erythroid ALAS2 expression is refractory to the presence of haem [4]. At least 22 mutations in the cDNA of the erythroid isoform ALAS2 have been reported in patients with X-linked sideroblastic anaemia [7]. This may provide a partial explanation for the development of abnormal porphyrin profile and photosensitivity seen in some patients with sideroblastic anaemia [8]. Genetic defects of ALAS1 have not been reported.
5-Aminolaevulinate Dehydratase
5-Aminolaevulinate dehydratase (ALAD), previously known as PBG synthase, condenses two molecules of ALA to form PBG [9]. Starting from this step, until the step of coproporphyrinogen oxidase, haem biosynthesis occurs in the cytoplasm of the cells (see Fig. 107.1). The activity of this cytosolic enzyme is up to 100-fold higher than ALA synthase, so all of the ALA synthesized is converted to PBG. This enzyme is encoded by a gene with 15,913 base-pairs, containing two non-coding exons (1A and 1B) and 11 coding exons. A housekeeping transcript and an erythroid-specific transcript have been identified, both encoding the same 330 amino acid protein [10]. ALA dehydratase has a molecular mass of 36 kDa; its gene has been located to chromosome 9p34 (see Table 107.2) [4,11].
Clinical Significance
Compound heterozygous deficiency of this enzyme is associated with ALA dehydratase deficiency porphyria (ADP), a rare form of porphyria, with only four patients reported (see Box 107.2 and Fig. 107.1) [12,13]. Heterogeneity in the gene mutation may explain the distinct phenotypes, ranging from a severely affected infant to an essentially asymptomatic 68-year-old man. One study reported a glycine-to-arginine substitution at residue 133 and a valine-to-methionine substitution at residue 275; another study reported an arginine-to-tryptophan substitution at position 240 and an alanine-to-threonine substitution at position 274 [14,15]. No photosensitivity has been reported in these patients.
Porphobilinogen Deaminase
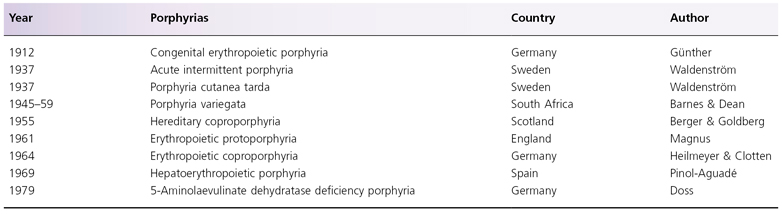
Porphobilinogen deaminase (PBGD) is also known as hydroxymethylbilane synthase. Four molecules of PBG, in the presence of PBGD, are converted into hydroxymethylbilane. Hydroxymethylbilane is a linear tetrapyrrole which, upon ring closure, forms the basic structure of the porphyrin molecule (see Fig. 107.2) [16]. A spontaneous, non-enzymatic hydroxymethylbilane ring closure results in the formation of uroporphyrinogen (urogen) I. Alternatively, urogen III can be formed in the presence of urogen III synthase (see below). These two isomers differ only in the D-ring of the tetrapyrrole, resulting in the switching of the positions of acetyl and propyl side-chains.
Human PBGD gene has 10,024 base-pairs, containing 15 exons and 14 introns [17]. Similar to ALAS, there are erythroid and non-erythroid isoforms of PBGD, with two distinct mRNA arising from two overlapping transcription units of a single gene, which is located on chromosome 11 (see Table 107.2) [18,19]. The erythroid isoform is encoded by the unit located 3 kb downstream from the other, which encodes for the non-erythroid isoform. Human erythroid-specific PBGD has 344 amino acids, and the non-erythroid specific isoform has 361 amino acid residues. After ALAS, this enzyme is present in the lowest concentration among enzymes in the haem biosynthetic pathway.
Clinical Significance
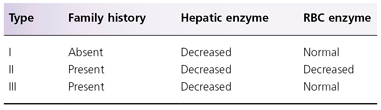
Deficiency of PBGD is responsible for acute intermittent porphyria (AIP) (see Box 107.2 and Fig. 107.1) [4,20,21]. Over 170 mutations in the PBGD gene isolated from patients with AIP have been reported [22–24]. Over 50% of Swiss AIP patients carried the W283X mutation, and one-third of patients in Holland had the R116W mutation. No phenotypic–genotypic correlation has been observed in AIP. Based on pattern of mutations, AIP has been classified into three types. Type I has cross-reactive immunological material (CRIM)-negative PBDG mutations, resulting in reduced enzyme activity and protein concentration (the latter is due to the inability of the abnormal protein to be detected by immunological methods). Type II consists of patients with mutations resulting in a 50% decrease in enzyme activity in non-erythroid tissue but normal erythroid activity (also known as AIP variant). Type III has CRIM-positive mutations with decreased enzyme activity and structurally abnormal protein. The enzyme deficiency results in accumulation of PBG and ALA, both of which are non-photosensitizing compounds; thus, patients with AIP do not have photosensitivity. Accumulation of PBG is the basis for the Watson–Schwartz and Hoesch tests [25]. A study of 196 Finnish AIP patients and 45 family members showed that measurement of urinary PBG identified all patients during an acute attack and 85% of patients during remission; therefore, urinary PBG measurement is considered the best biochemical test for AIP [21].
Uroporphyrinogen III Synthase
Uroporphyrinogen III synthase (UROS), previously known as urogen III co-synthase or hydroxymethylbilane hydrolyase, catalyses the ring closure of the linear hydroxymethylbilane molecule to urogen III, a tetrapyrrole. Its gene is located on chromosome 10; it has an approximate genome size of 34 kb (see Table 107.2). The gene has two promoters that generate housekeeping and erythroid-specific transcript with unique 5′-untranslated sequences (exons 1 and 2A), followed by nine common coding exons (2B to 10) [26]. The housekeeping transcript is present in all tissues, whereas the erythroid transcript is found only in erythropoietic tissues. The enzyme has 265 amino acid residues and an estimated molecular mass of 28.6 kDa [4,27,28].
Clinical Significance
A deficiency of this enzyme is associated with congenital erythropoietic porphyria (CEP, Günther disease), one of the most mutilating forms of porphyria, with an onset in infancy or early childhood, although milder late-onset CEP has been reported in a few patients [29,30]. CEP is characterized biochemically by elevated levels of type I isomer (see Table 107.2) [26]. At least 35 mutations of the UROS gene have been reported in patients with CEP [29,31–33]. The Cys73Arg (C73R) mutation is the most common. Correlation between mutations and phenotypic expressions has been observed in CEP, with C73R/C73R homozygotes having the most severe form of the disease [29].
Uroporphyrinogen Decarboxylase
Uroporphyrinogen decarboxylase (UROD) catalyses the sequential decarboxylation of both type I and III isomers of urogen (an 8-carboxyl porphyrinogen), to 7-, 6-, 5- and, eventually, 4-carboxyl porphyrinogen; the last is also known as coproporphyrinogen (coprogen). Subsequent enzymatic steps convert type III isomers into haem; in contrast, conversion of type I isomers stops with the formation of coprogen I.
The gene for human urogen decarboxylase is located on chromosome 1, is over 3 kb in length and has two initiation sites and 10 exons [34]. The enzyme has a molecular mass of 41 kDa and 367 amino acid residues. In contrast to the first four enzymes discussed in this chapter, no erythroid-specific isoform of UROD has been described.
Clinical Significance
Defective UROD is responsible for the development of porphyria cutanea tarda (PCT) and hepatoerythropoietic porphyria (HEP), considered to be a homozygous form of familial PCT (see Table 107.3 and Fig. 107.1) [35–38]. Enzyme activity is approximately 50% of normal in PCT and 10% of normal in HEP. The most common type of PCT, the sporadic or type I PCT, is associated with decreased enzyme activity in the liver (Table 107.4); type I PCT is not seen in children. Familial PCT is divided into two types; both are associated with a decreased hepatic UROD activity [35,36]. One type is also associated with a decreased enzyme activity in erythrocytes; this is known as type II PCT. Erythrocyte enzyme activity is normal in the other type of familial PCT, known as type III PCT. The substrates for UROD are hydrophilic porphyrinogens with relatively large numbers of carboxyl side-chains, which account for the elevated levels of urinary porphyrins in these patients (see Table 107.3).
Table 107.3 Porphyrin profiles of childhood porphyrias
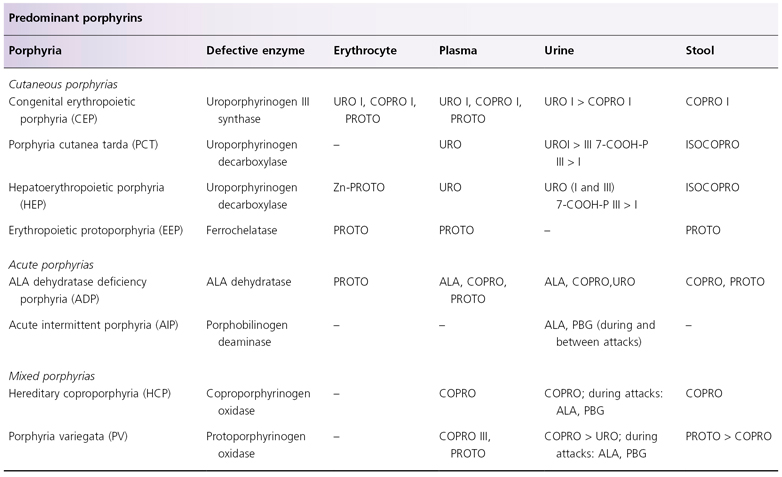
ALA, 5-aminolaevulinic acid; 7-COOH-P, 7-carboxyl porphyrin; COPRO, coproporphyrin; ISOCOPRO, isocoproporphyrin; PBG, porphobilinogen; PROTO, protoporphyrin; URO, uroporphyrin.
Table 107.4 Types of porphyria cutanea tarda
Mutations of the UROD gene have been detected in type II PCT and in HEP [38]. At least 65 mutations of UROD genes have been reported, four of them causing both HEP and familial PCT [39–43]. Heterogeneity in the UROD gene mutation occurs in familial PCT and in HEP. In HEP, this includes a glutamic acid-to-lysine substitution at 167, an arginine-to-glycine substitution at 292 and a glycine-to-glutamic acid substitution at 281. In the case of this last mutation, it was demonstrated that HEP is a homozygous form of familial PCT [43]. In contrast to familial PCT, analysis of UROD cDNA in sporadic PCT showed that the UROD locus is normal [44].
Adult patients with type I and type II PCT are associated with multiple risk factors, including exposure to alcohol and oestrogen, mutation in the HFE (hereditary haemochromatosis) gene, hepatitis C virus infection and human immunodeficiency virus infection [38]. These associations have not been reported in children. However, precipitation of clinical HEP in a 2-year-old child by hepatitis A infection has been reported [44].
Coproporphyrinogen Oxidase
Coproporphyrinogen oxidase (CPO) catalyses the sequential removal of the two carboxyl groups and the two hydrogen atoms from 4-carboxyl porphyrinogen (coprogen) III to harderoporphyrinogen (harderogen), a 3-carboxyl porphyrinogen, and to protoporphyrinogen (protogen), a 2-carboxyl porphyrinogen (see Fig. 107.1). This is the step in which haem biosynthesis returns from the cytosol into the mitochondrion. This enzyme is located at the outer surface of the inner mitochondrial membrane. Its cDNA has an open reading frame (ORF) of 1062 base-pairs, encoding a protein of 354 amino acid residues with a molecular mass of 40.3 kDa. The mature protein consists of 323 amino acid residues with a putative leader peptide of 31 amino acids; it has a molecular mass of 36.8 kDa [45]. The gene is located on chromosome 3 (see Table 107.2), with a genome size of 14 kb, and consists of seven exons and six introns [46].
When UROD is defective, as in PCT, CPO catalyses the oxidative decarboxylation of 5-carboxyl porphyrinogen to form dehydroisocoproporphyrinogen (see Fig. 107.1). The latter can undergo spontaneous non-enzymatic hydration to isocoproporphyrinogen (isocoprogen); the oxidation product of the latter, isocoproporphyrin, is characteristically elevated in the stool of patients with PCT. Alternatively, UROD may catalyse the decarboxylation of dehydroisocoproporphyrinogen to form the 3-carboxyl porphyrinogen harderoporphyrinogen, thus re-entering the normal pathway of haem biosynthesis.
Clinical Significance
Decreased activity of CPO is associated with hereditary coproporphyria, characterized biochemically by elevated levels of urinary and faecal coproporphyrin, predominantly of the III isomer; urinary ALA and PBG levels are also frequently elevated (see Table 107.3) [46,47]. At least 37 mutations and polymorphisms of the CPO gene have been reported [46,48]. As described above, this enzyme plays a significant role in the characteristic elevation of stool isocoproporphyrin seen in PCT.
Protoporphyrinogen Oxidase
Similar to the oxidation of other porphyrinogens to their corresponding porphyrins, oxidation of protogen to protoporphyrin can occur spontaneously. However, for haem biosynthesis to proceed at a normal rate, the oxidation of protogen to protoporphyrin is catalysed by protoporphyrinogen oxidase (PPO). This enzyme is located in the inner mitochondrial membrane and consists of 477 amino acids with a molecular mass of 50.8 kDa. Its gene was cloned in 1995; it has 13 exons and spans 5.5 kb [49,50].
Clinical Significance
Defective PPO is seen in porphyria variegata (PV) (see Table 107.3 and Fig. 107.1) [50]. At least 128 mutations of the PPO gene have been reported in PV [51–53]. The R59W mutation is present in >95% of PV patients in South Africa, representing the effect of the founder gene [50]. No correlation has been noted between phenotype and type of mutations. Rare cases of homozygous PV have been reported; most were actually compound heterozygous for defects in the PPO gene [54].
Defective PPO accounts for the observation that, in patients with PV, in the urine the level of coproporphyrin is greater than the level of uroporphyrin, and in the stool the level of protoporphyrin is greater than the level of coproporphyrin. In contrast, the reverse is observed in PCT, which may have similar cutaneous manifestations as PV. This is because the defective enzyme in PCT catalyses a series of reactions, starting with one that occurs six steps before the involvement of PPO, resulting in marked elevation of urinary uroporphyrin and the presence of faecal isocoproporphyrin (see Table 107.3 and Fig. 107.1).
During acute attacks, ALA and PBG concentrations are elevated. In vitro, coproporphyrinogen III and protoporphyrinogen IX, both elevated in PV, have been shown to suppress the activity of PBGD. Therefore, it has been postulated that the acute attacks in PV are caused by an inhibition of PBGD by these porphyrinogens [50]. It should be noted that, although symptomatic patients will always have an abnormal porphyrin profile, many asymptomatic PV patients may have no detectable abnormality in the porphyrin profile.
Ferrochelatase
Ferrochelatase, also known as haem synthase, is located in the inner membrane of the mitochondrion. It catalyses the insertion of ferrous iron (Fe2+), but not ferric iron (Fe3+), into protoporphyrin to form haem, which diffuses out from the mitochondrion into the cytoplasm. Its gene, which has been assigned to chromosome 18, contains 11 exons and 10 introns, and spans 45,000 base-pairs [55,56]. Its cDNA has an ORF of 1269 base-pairs, encoding a protein with 423 amino acid residues [57]. The mature protein has a molecular mass of 40 kDa.
Clinical Significance
Defective ferrochelatase is found in erythropoietic protoporphyria (EPP) (see Table 107.2 and Fig. 107.1) [58,59]. The lipophilic nature of the substrate of ferrochelatase, protoporphyrin, accounts for the finding that urinary porphyrin levels are normal in patients with EPP (see Table 107.3). At least 78 point mutations, base-pair deletions and base-pair insertions have been reported in the ferrochelatase gene in EPP [60–63]. Symptomatic EPP is associated with <50% of enzyme activity, whereas asymptomatic carriers only have mild enzyme deficiency. This can be explained by the suggestion that manifestations of photosensitive EPP require the presence of a ferrochelatase defect (from a biochemically abnormal but asymptomatic parent), and co-inheritance of a variant of normal ferrochelatase gene with reduced gene expression (present in about 10% of the general population) [64,65]. Compound heterozygosity for defective ferrochelatase gene is associated with liver failure [59].
Pathogenesis
Cutaneous Porphyrias
Reactive oxygen species, inflammatory mediators and inflammatory cells have been shown to contribute to the development of cutaneous lesions in porphyrias [66]. Upon exposure to Soret band (400–410 nm) radiation, ‘excited state’ porphyrins are generated, which can interact with the oxygen molecules to form singlet oxygen (1O2). Superoxide anion (O2−), hydroxyl radical (OH) and hydrogen peroxide (H2O2) can also be generated. In vitro, the reactive oxygen species and the peroxides are damaging to the cell membrane, resulting in mediator release from mast cells, damage to hepatic and epidermal microsomal cytochrome P450 and damage to lysosomal and mitochondrial membranes [66]. In a mouse model, superoxide anions have been shown to contribute to the development of haematoporphyrin-induced phototoxicity [67]. Clinically, β-carotene, a scavenger of singlet oxygen, is used to treat the cutaneous symptoms of EPP [68].
Mast cells, neutrophils and the complement system also participate in porphyrin-induced phototoxicity. In vitro, protoporphyrin, but not uroporphyrin, has been shown to induce mast cell mediator release in the presence of Soret band radiation [69]. The reason for the differential effect is most likely the fact that protoporphyrin is lipophilic, whereas uroporphyrin is hydrophilic. This effect may account for the sunburn-like manifestations of porphyrias presenting with the acute syndrome (CEP, HEP and EPP); all of them are associated with elevated protoporphyrin (see Box 107.1 and Table 107.3). The lack of effect of uroporphyrin on mast cells may partly account for the absence of the acute manifestations in PCT. In vitro, protoporphyrin and radiation are known to cause membrane damage to neutrophils [70]. Activation of the complement system by porphyrin and radiation may contribute to the development of the phototoxicity [71]. In animal models, porphyrin-induced phototoxicity is associated with release of mast cell mediators and is suppressed by antihistamine and in mast cell-deficient, complement-deficient or neutropenic animals [66]. The role of mast cells in porphyrin-induced phototoxicity is further supported by the observation that terfenadine inhibits the immediate flare reaction in patients with EPP [72].
Uroporphyrin has been shown to increase collagen synthesis by fibroblasts in a radiation-independent fashion [73]. This may account for the presence of sclerodermoid skin lesions seen in both sun-exposed and sun-protected skin of patients with PCT.
The cause of the mottled periorbital pigmentary changes and hypertrichosis is not known. The mechanism for skin fragility and vesicle formation in subacute cutaneous porphyrias is also not completely understood. It is possible that C3a and C5a generated by porphyrin-induced photoactivation of the complement system in turn induce release of proteases from cutaneous mast cells. Mast cell-derived proteases have been shown to be able to cleave the dermoepidermal junction at the lamina lucida level.
Acute Porphyrias
It is known that damage to the autonomic and peripheral nervous system accounts for the abdominal and neurological symptoms seen in these patients. As all patients with acute attacks have elevated ALA and PBG, it has been postulated that these compounds are neurotoxic [74]. Alternatively, it is possible that abnormal haem biosynthesis in the neural tissue contributes to its dysfunction [75].
Mixed Porphyrias
The pathogenesis of signs and symptoms of mixed porphyrias is identical to that of cutaneous porphyrias and acute porphyrias.
Pathology.
Histologically, vesiculobullous lesions of PCT represent a dermoepidermal splitting without dermal inflammatory reaction. Dermoepidermal separation occurs in the lamina lucida [76]. The dermal papillae profile is preserved and a ‘festooned’ aspect of the blister floor is characteristic.
In the chronic phase of erythropoietic protoporphyria, the skin shows the presence of abundant amorphous hyaline eosinophilic material around the superficial blood capillary walls and in the upper dermis. This dense material is periodic acid–Schiff (PAS) positive and diastase resistant. In young children, when the disease is evolving for a short time, the first objective chronic manifestation may only be a thin PAS-positive rim around the upper dermal vessels [77]. Ultrastructural studies show a reduplication of the basement membrane of the upper dermal blood capillaries [78].
Clinical Features
Incidence of Porphyrias in Children
The incidence of the porphyrias varies from country to country. The estimated incidence of porphyria cutanea tarda (PCT) in the USA is 1 in 25,000 [2], and in the former Czechoslovakia 1 in 5000. The incidence of PV in the white population of South Africa is 1% [79]. EPP is very rare in some countries but more common in others; in the UK the estimated prevalence is 1 in 130,000 (382 cases were identified in the UK between 1979 and 1985) [80].
In the review of 793 cases of porphyria in the Department of Dermatology of the Hospital Clinic of Barcelona from 1969 to 1994, only 21 were children. Six had PCT, eight had EPP, three had CEP and four had HEP [81]. No cases of acute or mixed porphyria were seen in children. However, these figures refer only to the age of diagnosis, and mild childhood symptoms may have been ignored in some patients.
Porphyria cutanea tarda is significantly more common in adults than in children. A study of patients with PCT at the Hospital Clinic of Barcelona revealed that 87.5% had type I PCT, which is sporadic and develops during adulthood. The familial type II and III forms, which may begin during childhood, were seen in 10% and 2.5% of patients respectively [36].
Cutaneous Porphyrias
Cutaneous porphyrias are the most common in children [82]. Dermatological manifestations appear in two different forms.
1 Acute cutaneous syndrome is an acute photosensitivity that resembles sunburn. The intensity of symptoms is not proportional to the duration of exposure to sunlight or, in some cases, to artificial light sources. This clinical presentation is very characteristic of EPP, in which there is an overproduction of protoporphyrin (a lipophilic porphyrin). However, acute photosensitivity may also develop in homozygous porphyrias when production of hydrophilic porphyrins is extremely high. This phenomenon is seen in CEP and HEP.
2 Subacute cutaneous syndrome
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


