Introduction
Langerhans cell histiocytosis
Non-Langerhans cell histiocytosis
Haemophagocytic lymphohistiocytosis
Introduction
The histiocytic disorders (HD) comprise a wide variety of conditions characterized by the proliferation, infiltration and accumulation of histiocytes in the body tissues. The course of these conditions is variable, ranging from complete spontaneous resolution to progressive, multisystem, potentially lethal disease [1,2]. Classically, the histiocytoses have been referred to as one of the ‘great mimickers’ since they are commonly confused with other conditions.
Box 103.1 Classification of Histiocytic Disorders
Disorders of Varying Biological Behaviour
Dendritic Cell Related
- Langerhans cell histiocytosis
- Juvenile xanthogranuloma and related disorders including:
Erdheim-Chester disease
Solitary histiocytomas with juvenile xanthogranuloma phenotype
- Secondary dendritic cell disorders
Monocyte/Macrophage Related
- Haemophagocytic lymphohistiocytosis
Familial and sporadic
- Secondary haemophagocytic syndromes:
Infection associated
Malignancy associated
Autoimmune associated
Other
- Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease)
- Solitary histiocytoma of macrophage phenotype
Dendritic Cell Related
- Histiocytic sarcoma (localized or disseminated)
- Monocyte/macrophage related
- Leukaemias:
Monocytic leukaemias M5A and B
Acute myelomonocytic leukaemia M4
Chronic myelomonocytic leukaemias
- Extramedullary monocytic tumours or sarcoma (monocytic counterpart of granulocytic sarcoma)
- Macrophage-related histiocytic sarcoma (localized or disseminated)
Adapted from Favara et al. Med Pediatr Oncol 1997;29(3):157–66.
Cellular Origin
Several cell lines are implicated in the different types of histiocytoses. Identification of the predominant histiocyte type is important for a correct diagnosis.
In the development cycle, the CD34+ stem cell is the origin of the majority of histiocytes. This stem cell matures into CD14- or CD14+ histiocytes, depending on the influence of its microenvironment. The CD14- cells, in the presence of tumour necrosis factor α and granulocyte-macrophage colony-stimulating factor, may evolve into Langerhans cells (LC) and CD14+ cells, depending on the cytokines they are exposed to, differentiate into interstitial dermal dendrocytes (also referred to as interstitial dendritic cells) [3] or into monocytes/macrophages.
Histiocytic disorders can involve all three cell types, and can therefore be divided into Langerhans cell histiocytosis (LCH), when the LC are affected, non-Langerhans cell histiocytosis (non-LCH), when the affected line is the interstitial dermal dendrocyte, and haemophagocytic lymphohistiocytosis (HLH), when the macrophages are involved.
References
1 Haupt R, Nanduri V, Calevo MG et al. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society–Late Effects Study Group. Pediatr Blood Cancer 2004;42(5):438–44.
2 Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer 2007;48(5):555–60.
3 Banchereau J, Fay J, Pascual V, Palucka AK. Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Novartis Found Symp 2003;252:226–35; discussion 35–8, 57–67.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis is the most common of the HD in childhood, affecting 2–5 million children yearly [1].
History.
The first description of a 4-year-old patient with impetigo and three lytic lesions in the calvarium was given by Smith in 1865 [2]. In 1868, Paul Langerhans first described the non-pigmentary dendritic cells that now carry his name. Alfred Hand, Artur Schüller and Henry Christian, in 1893, 1915 and 1920 respectively, described a condition with exophthalmos, skull defects and diabetes insipidus known as Hand–Schüller–Christian disease. A few years later, Siwe put together several reports of patients with organomegaly, lymphadenopathy, localized tumours in the bones, secondary anaemia and haemorrhagic tendency as well as hyperplasia of non-lipid storing macrophages, that included a case by Letterer; the co-existence of these findings was referred to as Letterer–Siwe disease. In the 1940s, a description of eosinophilic granuloma of the bone was given. In 1941, Faber noted that all these conditions were variations of the same disease. In 1953, Lichtenstein grouped these three conditions under the term histiocytosis X [3] and later, in 1973, Nezelof showed that these were the result of proliferation of LCs [4]. In 1961, Birbeck et al. described the characteristic Birbeck granules seen in electron microscopy that help identify LC [5]. A few years later, in 1973, Hashimoto and Pritzker described the entity congenital self-healing reticulohistiocytosis [6]. Later, advances in immunological and ultrastructural studies that revealed the relationship between all of these conditions formed the basis for the proposed classification by the Writing Group of the Histiocyte Society in 1987 that grouped all of these under the term ‘Langerhans cell histiocytoses’ [7].
Epidemiology.
Langerhans cell histiocytosis, although rare, is the most common of the HDs in childhood, occurring in 2.2–8.9 children per million per year [8-11]. Children under 1 year of age account for the largest proportion of the affected population (9.0–15.3 cases per million per year) [8,11]. HD frequency rates significantly decrease with age (4.6, 1.1, 0.7 cases per million per year in 1–4, 5–9 and 10–14 year olds respectively) [8], and about 2% LCH cases present in the neonatal period (before 28 days of age) [12].
A male predominance has been reported, with boys affected twice as often as girls [13]. No racial or geographic predilection has been observed. It is considered a non-hereditary disorder, although cases of twins and siblings have been described [14]. Seasonal variation was suggested, with cases being more frequent during autumn and winter [9], and in association with climate changes, [15] but this has not been duplicated [8,16].
Pathogenesis.
The underlying aetiology of LCH remains unclear. An infectious trigger such as Epstein–Barr virus and HHV-6 initiating LC proliferation has been proposed [17-19], but was not consistently found in all studies. Due to evidence of clonality in the cell lines, it was also suggested that LCH might be a neoplastic growth [20-23]. In addition, a relationship between LCH and myelodysplastic syndromes was proposed [24]. However, the most recent genetic testing is more in keeping with LCH being the result of a restricted oligoclonal stimulation rather than of an unlimited neoplastic proliferation [25].
Another proposed mechanism is an aberrant immune response reaction to common pathogens, due to congenital predisposition of the host’s immune system [26], leading to an excessive inflammatory process. Some studies have also demonstrated an imbalance in the chemokine expression (‘cytokine storm’) that regulates LCH [27] and, more recently, IL-17 dysregulation has been implicated in this condition [28]. The lack of genomic aberrations in LCH led to speculations that epigenetic events may play an important role in LCH [25].
Classification.
In 1987, the Writing Group of the Histiocyte Society attempted to classify these conditions according to their relationship to normal histiocyte subsets [7]. This classification was revised, and in 1997 Favara et al. published a new classification taking into consideration the biological behaviour of these disorders, making a separation between the truly malignant and the ones with varying biological behaviour (Box 103.1). These two broad categories were broken down depending on whether they belong to the dendritic cell or macrophage/monocytes pathways [29].
Depending on the clinical presentation, the LCH can be separated into an acute, disseminated form (disseminated LCH with haematological dysfunction, formerly Letterer–Siwe disease); a chronic, localized form (eosinophilic granuloma); a progressive, multifocal, chronic form (initially called Hand–Schüller–Christian disease); and a benign, self-healing form (congenital self-healing reticulohistiocytosis, previously called Hashimoto–Pritzker disease). Many believe that these clinical entities overlap with each other, and therefore they should be considered within a common clinical spectrum.
Clinical Features.
Clinical manifestations of LCH can be unifocal or single-system (S-S) disease or multifocal or multisystem (M-S) disease. Apart from the skin and bone, other sites can be affected including lymph nodes, central nervous system, spleen, liver and thymus.
Cutaneous manifestations of LCH are variable (papules, pustules, vesicles, ulcerated or non-ulcerated nodules, petechiae, etc.) and may mimic more benign skin conditions (e.g. seborrhoeic dermatitis) (Fig. 103.1). Its protean clinical picture may delay the diagnosis. In children with LCH presenting from birth to 4 weeks of age, the diagnosis was not confirmed until they were 3.5 months old [30]. Its non-specific nature in addition to the common involvement of the skin in up to 89% of cases of S-S disease [11] and 75–100% of cases of M-S disease [31] raises the importance of considering LCH in the differential diagnosis of persistent skin rashes.
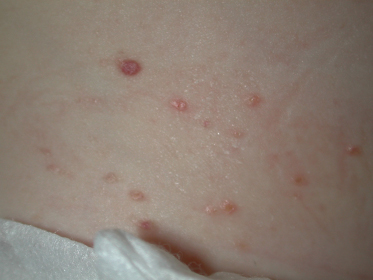
The bone may also be the primary disease site. Some studies report the bone as the site most commonly affected [32] in up to 80% of cases [33]. In S-S disease bone involvement seems to be as high as 85%, and is up to 74% in M-S disease [34,35]. While the bones of the skull and face are most frequently involved, lytic lesions may also be found in the flat bones such as ribs, clavicle and scapula. Mastoid involvement mimics mastoiditis, and mandibular and maxillar bone destruction may lead to a ‘floating teeth’ appearance on the radiographs [36].
Haematological findings vary from long-standing anaemia to severe pancytopenia. Bone marrow involvement is rare, and occurs late in the course of the disease [37]. Some of the haematological findings, such as anaemia, thrombocytopenia, leucopenia or pancytopenia, may result from bone marrow involvement.
Pulmonary findings are seen in 12–23% of cases [38,39]. Patients can clinically manifest with respiratory symptoms such as cough, thoracic pain and tachypnoea. Lung LCH can present as interstitial infiltrates, cystic nodules and later in the course of the disease as pneumothorax (due to bulla formation and rupture) or as emphysematous changes with diffuse fibrosis.
Hepatomegaly, splenomegaly and lymphadenopathy are also manifestations of LCH. Involvement of the liver and spleen can be seen in 15–50% of cases [38,39]. Initial hepatic lesions may progress to severe fibrosis, biliary cirrhosis and liver failure [40]. The spleen involvement may contribute to thrombocytopenia.
Diabetes insipidus is the most common endocrinological finding, followed by growth hormone deficiency secondary to pituitary gland involvement [41] and involvement of the pancreas, gonads and thyroid. Thyroid involvement, the result of histiocytic infiltration, leads to hypothyroidism, progressive goitre and upper airway obstruction [42,43]. LCH can also be associated with gastrointestinal symptoms, which can manifest as haematochezia, non-bloody diarrhoea, perianal fistulas or constipation [44].
Central nervous system involvement is rare in LCH; progressive intellectual deficit, encephalopathy, spastic paraplegia and tetraplegia, ataxia and blurred vision have been described [38].
Otological changes of LCH can resemble those of acute and chronic ear infections; a biopsy is needed to make a definitive diagnosis [45].
Reports of LCH presenting in the neonatal period with purplish or violaceous-reddish, firm skin lesions led to the inclusion of this disorder in the differential of a ‘blueberry muffin baby’ [46].
Eosinophilic Granuloma
The chronic localized form or eosinophilic granuloma (EG) is the most common in children, accounting for about 70–80% of paediatric LCH [33,47]. It is a benign, tumour-like condition in which clonal proliferation of Langerhans cells is identified. About 90% of patients with this condition are between the ages of 5 and 15 years [48] with a male predominance [49]. Bone lesions are seen in about 90% of cases [50] and present as a painless, destructive, bone lesion. Unifocal involvement is more common than the multifocal form, presenting in 64.3% of cases [51]. The flat bones (skull, pelvis, vertebrae, mandible and ribs) are involved in 70% of cases. In about 43–80% of cases the skull is involved [47] and although unifocal involvement is more common, multifocal lesions are seen in about 20% of patients with skull involvement [47]. The long bones are involved in 30% of cases [52], with femur, humerus and metaphases more commonly affected [48].
Two phases of EG in the bone have been described: an acute phase in which the lesion is destructive, osteolytic and has poor margins, and a chronic phase, when it becomes well defined and easily confused with the radiological findings of chronic osteomyelitis or a benign bone tumour [53]. Less frequently, EG can affect the soft tissues and orbits, present as torticollis [48,49,54,55] and exceptionally as a nodulo-ulcerative lesion [50].
Disseminated LCH with Haematological Dysfunction
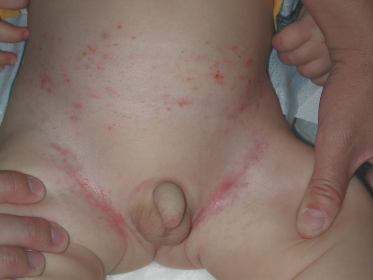
The former Letterer–Siwe disease, currently known as disseminated LCH with haematological dysfunction (DLCH), accounts for less than 15% of cases of LCH [56] and typically affects children under the age of 2 years. In addition to mucocutaneous lesions, it presents with hepatosplenomegaly, lymphadenopathy, fever, anaemia and thrombocytopenia. The skin lesions may be one of the first manifestations of DLCH, and present as a ‘seborrhoeic dermatitis’-like eruption involving the nappy area, hairline and behind the ears (Fig. 103.2). Petechiae and purpura are common, and occasionally extensive ulcerations and suprainfection of lesions may also be found. This form of LCH usually carries a worse prognosis and is unlikely to resolve on its own. Paediatric patients with this type of LCH have been reported to relapse many years after the initial presentation manifesting as other types of LCH, and thus long-term follow-up is recommended [57].
Fig. 103.2 ‘Seborrheic dermatitis’-like eruption involving the diaper area in a patient with disseminated LCH with haematological dysfunction.
Progressive, Multifocal, Chronic Form (Hand–Schüller–Christian Disease)
Classically, Hand–Schüller–Christian disease (HSCD) was described as the triad of exophthalmos, diabetes insipidus and osteolytic lesions of the skull. Currently, HSCD is known as a progressive, multifocal, chronic form of LCH (PMC-LCH). The most typical findings in PMC-LCH are the lytic lesions of the bones, but the presence of exophthalmos and diabetes insipidus as well as cutaneous findings have been reported. Not all the findings need to be present in order to make the diagnosis. The age of onset is usually between 2 and 6 years of age. Skin lesions can present as a seborrhoeic dermatitis-like form or as yellow-brown nodules and tumours. The exophthalmos is due to the accumulation of LC in the retro-orbital area. Diabetes insipidus has been described months to years after the initial presentation [30]. Premature tooth eruption may be the first manifestation of this disease [58].
Congenital Self-Healing Reticulohistiocytosis
Hashimoto and Pritzker first described this entity in 1973 and since then over 90 cases have been reported [59]. Congenital self-healing reticulohistiocytosis (CSHRH) is characterized by early-onset skin lesions in an otherwise healthy infant, with no or minimal systemic involvement, in which histopathology shows a LC infiltrate and lesions spontaneously involute. Clinical features are classically present at birth or in the first months of life [60,61] and only one case has been reported in an 8-year-old girl [62].
The typical presentation is with multiple, disseminated, firm, red-brown, painless papules or nodules (Fig. 103.3). Less commonly, vesicles, pustules, crusts and haemorrhagic bullae can occur [63,64]. Approximately 25–30% of cases present as red-brown, firm nodules with a tendency to ulceration [59,65]. Lesions predominantly involve the scalp, face and trunk, but acral and mucosal involvement has also been described [60,63,66]. Lesions typically regress by 3 months of age with residual pigmentation.
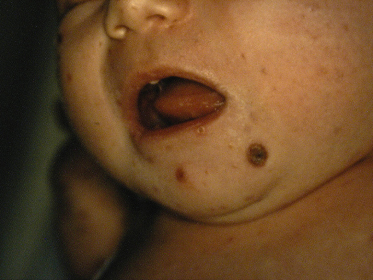
Although systemic involvement is generally absent, reports of transient haematological anomalies [67,68], hepatic abnormalities, gastrointestinal involvement [69], splenomegaly, palpable lymphadenopathy, bone [70] and lung [69,71] involvement have been described. The multisystem involvement does not indicate a worse prognosis [67,71]. The length of follow-up is not clear; relapses up to 4 years of age have been reported [72]. The relapses have been seen in the skin [68], mucosa [73], bone [68] and pituitary gland [72]. No treatment is needed for CSHRH due to its excellent prognosis.
Evaluation.
A complete clinical history and physical examination will raise the suspicion of LCH that needs to be pathologically confirmed. When skin lesions are not present, deeper tissue biopsies, such as bone biopsies or bone marrow aspirates, may be necessary. The initial investigations should always include a complete blood count with differential, liver function tests, coagulation studies, urine osmolality, chest radiograph, skeletal radiograph survey and abdominal ultrasound. Targeted endocrine testing as well as endoscopic evaluation of gastrointestinal tract, and pulmonary function tests can be useful when involvement of these systems is suspected.
While conventional radiographs reveal skeletal lesions, computed tomography may be necessary to further delineate lesions of the skull base, spine and pelvis [36]. Bone scintigraphy is not widely accepted for this condition, since about 20% of lesions may not be detected by bone scans [74]. MRI is most helpful for diagnosing lesions in the skull and central nervous system, but not specific for bone marrow involvement or soft tissue masses.
Pathology.
On light microscopy, the LCH is characterized by monomorphous, dense infiltrates of polygonal cells that have a pale cytoplasm with kidney-shaped nuclei. These cells are epidermotropic and present a pagetoid pattern. A few inflammatory cells, like lymphocytes, eosinophils, neutrophils and plasma cells, can be identified to a varying extent. It is also possible to detect polynucleated cells [6]. On histology alone, it is not possible to differentiate between the different types of LCH.
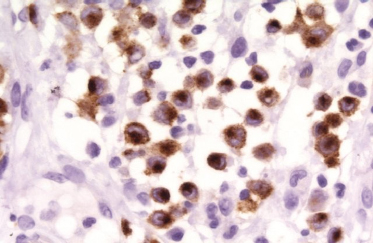
On immunohistochemical studies, LCH stains positively for S-100 and CD1a (Fig. 103.4). Antibodies that recognize epitopes of the LC-specific Birbeck granules currently identify these cells more precisely. The monoclonal antibody Lag (Langerhans cell-associated granulum) recognizes the intracellular antigen in the membrane of Birbeck granules and is used for cryopreserved material [75]. Langerin (CD207) is a monoclonal antibody against a transmembrane protein expressed in LC (Fig. 103.5), and it is highly specific and sensitive for detection of LC [76-79]. Langerin is found in the cell surface, and is a potent inducer of Birbeck granule formation.
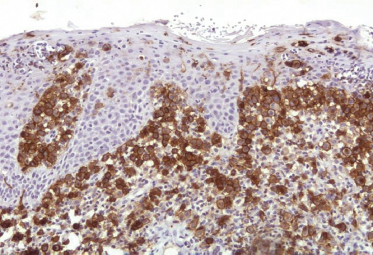
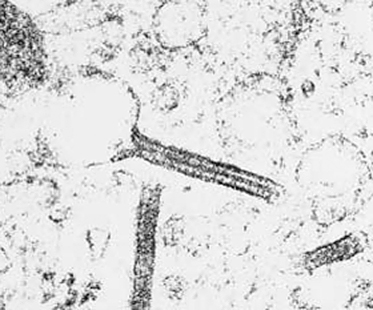
The presence of intracytoplasmic organelles (Birbeck granules) on electron microscopy is highly specific for LCH (Fig. 103.6). However, in CSHRH only 10–30% of histiocytic cells are found to have Birbeck’s granules [66].
Treatment.
In general, patients with S-S disease require minimum or no treatment, while M-S disease normally requires systemic interventions.
For persistent skin-only lesions, topical steroids or topical nitrogen mustard are useful [63,80,81]. Intralesional steroids or excision can also be used in solitary lesions [82]. Bone lesions respond to curettage, local irradiation, surgical resection, systemic or local corticoids, systemic chemotherapy or observation alone [54]. Due to a lack of randomized controlled trials comparing treatment options, there is no consensus on the optimal treatment modality.
When soft tissues are involved, or in M-S disease, cytotoxic drugs and systemic corticosteroids are required. About 50% patients requiring systemic treatment respond to steroid therapy alone [49]. Chemotherapeutic agents (vinblastine, methotrexate, chlorambucil and 6-mercaptopurine) are used when patients with M-S disease fail to respond to the first-line therapy. Refractory cases of LCH may require stem cell transplantation [83].
Prognosis.
In general, the prognosis of LCH depends on the extent of the LC infiltrates. The S-S disease has been reported to have excellent prognosis with survival of almost 100% of cases [34]. This seems to be true for patients with bone involvement alone, but seems to be less clear in cases of skin-only disease. Even in the bone-only disease, while survival is favourable, about 20–30% of these patients will have orthopaedic complications [34,84,85].
In S-S disease affecting only the skin, the outcome is less predictable; 30–56% of paediatric cases of skin-only LCH may progress to M-S disease [34,86,87]. Even in patients with skin-only congenital LCH, the progression rate to M-S disease has been estimated to be as low as 8% and as high as 60% [86]. In cases of EG, recurrences vary from 1.6% to 25% depending on the location of the lesions [49] but are typically associated with a positive outcome.
Children with M-S disease have significantly higher morbidity and mortality [88]. Approximately 40–65% of disease survivors suffer permanent sequelae [84,85,87,89]. Diabetes insipidus seems to be the most common permanent consequence seen in M-S disease, ranging from 35–60% of patients [84,85,87,90] to 7–20% of cases in the most recent studies [91].
Mortality rates for M-S disease have been estimated at between 20% and 50% [86]. Children younger than 2 years of age, with involvement of the risk organs (lung, liver, spleen or bone marrow) and non-responsive to treatment, have the worst prognosis [88,92-94]. A small subset of M-S disease may have spontaneous regression of lesions [95].
References
1 Lau LM, Stuurman K, Weitzman S. Skeletal Langerhans cell histiocytosis in children: permanent consequences and health-related quality of life in long-term survivors. Pediatr Blood Cancer 2008;50(3):607–12.
2 Coppes–Zantinga A, Egeler RM. The Langerhans cell histiocytosis X files revealed. Br J Haematol 2002;116(1):3–9.
3 Lichtenstein L. Histiocytosis X; integration of eosinophilic granuloma of bone, Letterer–Siwe disease, and Schuller–Christian disease as related manifestations of a single nosologic entity. AMA Arch Pathol 1953;56(1):84–102.
4 Nezelof C, Basset F, Rousseau MF. Histiocytosis X histogenetic arguments for a Langerhans cell origin. Biomedicine 1973;18(5):365–71.
5 Birbeck MS, Breathnach AS, Everall JD. An electron microscope study of basal melanocytes and high-level clear cells (Langerhans cells) in vitiligo. J Invest Dermatol 1961;37:51–63.
6 Hashimoto K, Pritzker MS. Electron microscopic study of reticulohistiocytoma. An unusual case of congenital, self-healing reticulohistiocytosis. Arch Dermatol 1973;107(2):263–70.
7 Chu A, D’Angio GJ, Favara BE et al. Histiocytosis syndromes in children. Writing Group of the Histiocyte Society. Lancet 1987;1(8526):208–9.
8 Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer 2007;48(5):555–60.
9 Stalemark H, Laurencikas E, Karis J, Gavhed D, Fadeel B, Henter JI. Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer 2008;51(1):76–81.
10 Muller J, Koos R, Garami M et al. [Experiences with Langerhans’ cell histiocytosis in children in Hungary]. Magy Onkol 2004;48(4):289–95.
11 Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer 2008;51(1):71–5.
12 Minkov M, Prosch H, Steiner M et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer 2005;45(6):802–7.
13 Sidler AK, Huston BM, Livasy C, Thomas DB. Pathological case of the month. Eosinophilic granuloma (Langerhans cell histiocytosis). Arch Pediatr Adolesc Med 2000;154(10):1057–8.
14 Arico M, Nichols K, Whitlock JA et al. Familial clustering of Langerhans cell histiocytosis. Br J Haematol 1999;107(4):883–8.
15 Chen RL, Lin KS, Chang WH et al. Childhood Langerhans cell histiocytosis increased during El Nino 1997–98: a report from the Taiwan Pediatric Oncology Group. Acta Paediatr Taiwan 2003;44(1):14–20.
16 Glass AG, Miller RW. U.S. mortality from Letterer–Siwe disease, 1960–1964. Pediatrics 1968;42(2):364–7.
17 Glotzbecker MP, Carpentieri DF, Dormans JP. Langerhans cell histiocytosis: a primary viral infection of bone? Human herpes virus 6 latent protein detected in lymphocytes from tissue of children. J Pediatr Orthop 2004;24(1):123–9.
18 Shimakage M, Sasagawa T, Kimura M et al. Expression of Epstein–Barr virus in Langerhans’ cell histiocytosis. Hum Pathol 2004;35(7):862–8.
19 Sakata N, Toguchi N, Kimura M, Nakayama M, Kawa K, Takemura T. Development of Langerhans cell histiocytosis associated with chronic active Epstein–Barr virus infection. Pediatr Blood Cancer 2008;50(4):924–7.
20 Bank MI, Rengtved P, Carstensen H, Petersen BL. Langerhans cell histiocytosis: an evaluation of histopathological parameters, demonstration of proliferation by Ki–67 and mitotic bodies. APMIS 2003;111(2):300–8.
21 Schouten B, Egeler RM, Leenen PJ, Taminiau AH, van den Broek LJ, Hogendoorn PC. Expression of cell cycle-related gene products in Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2002;24(9):727–32.
22 Willman CL, Busque L, Griffith BB et al. Langerhans’ cell histiocytosis (histiocytosis X) – a clonal proliferative disease. N Engl J Med 1994;331(3):154–60.
23 Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 1994;343(8900):767–8.
24 Egeler RM, Willman CL. Is Langerhans cell histiocytosis a myeloid dendritic stem cell disorder related to myelodysplastic disorders? Med Pediatr Oncol 2000;35(4):426–7.
25 Da Costa CE, Szuhai K, van Eijk R et al. No genomic aberrations in Langerhans cell histiocytosis as assessed by diverse molecular technologies. Genes Chromosomes Cancer 2009;48(3):239–49.
26 Arico M. Langerhans cell histiocytosis: too many cytokines, not enough gene regulation? Pediatr Blood Cancer 2006;47(2):118–19.
27 Annels NE, da Costa CE, Prins FA, Willemze A, Hogendoorn PC, Egeler RM. Aberrant chemokine receptor expression and chemokine production by Langerhans cells underlies the pathogenesis of Langerhans cell histiocytosis. J Exp Med 2003;197(10):1385–90.
28 Coury F, Annels N, Rivollier A et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med 2008;14(1):81–7.
29 Favara BE, Feller AC, Pauli M et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997;29(3):157–66.
30 Stein SL, Paller AS, Haut PR, Mancini AJ. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med 2001;155(7):778–83.
31 Hamre M, Hedberg J, Buckley J et al. Langerhans cell histiocytosis: an exploratory epidemiologic study of 177 cases. Med Pediatr Oncol 1997;28(2):92–7.
32 Yu RC, Chu AC. Lack of T-cell receptor gene rearrangements in cells involved in Langerhans cell histiocytosis. Cancer 1995;75(5):1162–6.
33 Azouz EM, Saigal G, Rodriguez MM, Podda A. Langerhans’ cell histiocytosis: pathology, imaging and treatment of skeletal involvement. Pediatr Radiol 2005;35(2):103–15.
34 Titgemeyer C, Grois N, Minkov M, Flucher-Wolfram B, Gatterer-Menz I, Gadner H. Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL-HX 83- and 90-study. Med Pediatr Oncol 2001;37(2):108–14.
35 Gadner H, Heitger A, Grois N, Gatterer-Menz I, Ladisch S. Treatment strategy for disseminated Langerhans cell histiocytosis. DAL HX-83 Study Group. Med Pediatr Oncol 1994;23(2):72–80.
36 Herman TE, Siegel MJ. Langerhans cell histiocytosis: radiographic images in pediatrics. Clin Pediatr (Phila) 2009;48(2):228–31.
37 McClain K, Ramsay NK, Robison L, Sundberg RD, Nesbit M Jr. Bone marrow involvement in histiocytosis X. Med Pediatr Oncol 1983;11(3):167–71.
38 Group TFLsCHS. A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group. Arch Dis Child 1996;75(1):17–24.
39 Rivera-Luna R, Martinez-Guerra G, Altamirano-Alvarez E et al. Langerhans cell histiocytosis: clinical experience with 124 patients. Pediatr Dermatol 1988;5(3):145–50.
40 Pirovino M, Jeanneret C, Lang RH, Luisier J, Bianchi L, Spichtin H. Liver cirrhosis in histiocytosis X. Liver 1988;8(5):293–8.
41 Dean HJ, Bishop A, Winter JS. Growth hormone deficiency in patients with histiocytosis X. J Pediatr 1986;109(4):615–18.
42 Diamond FB Jr, Shulman DI, Lacson A, Casadonte J, Favara B. Atypical dendritic cell-related histiocytosis with goiter and primary hypothyroidism. J Pediatr 1998;132(2):357–60.
43 Burnett A, Carney D, Mukhopadhyay S, Scalzetti EM, Leino D, Souid AK. Thyroid involvement with Langerhans cell histiocytosis in a 3-year-old male. Pediatr Blood Cancer 2008;50(3):726–7.
44 Hait E, Liang M, Degar B, Glickman J, Fox VL. Gastrointestinal tract involvement in Langerhans cell histiocytosis: case report and literature review. Pediatrics 2006;118(5):e1593–9.
45 Skoulakis CE, Drivas EI, Papadakis CE, Bizaki AJ, Stavroulaki P, Helidonis ES. Langerhans cell histiocytosis presented as bilateral otitis media and mastoiditis. Turk J Pediatr 2008;50(1):70–3.
46 Sankilampi U, Huikko-Tarvainen S, Karja V, Pirinen E, Naukkarinen A, Hollmen A. Congenital Langerhans cell histiocytosis mimicking a ‘blueberry muffin baby’. J Pediatr Hematol Oncol 2008;30(3):245–8.
47 Lee YS, Kwon JT, Park YS. Eosinophilic granuloma presenting as an epidural hematoma and cyst. J Korean Neurosurg Soc 2008;43(6):304–6.
48 Per H, Koc KR, Gumus H, Canpolat M, Kumandas S. Cervical eosinophilic granuloma and torticollis: a case report and review of the literature. J Emerg Med 2008;35(4):389–92.
49 Ulivieri S, Oliveri G, Filosomi G. Solitary Langerhans cell histiocytosis orbital lesion: case report and review of the literature. Neurocirugia (Astur) 2008;19(5):453–5.
50 Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, Gonzalez-Lopez MA. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. A lesion that may cause diagnostic error. J Cutan Pathol 2009;36(10):1109–13.
51 Lieberman PH, Jones CR, Steinman RM et al. Langerhans cell (eosinophilic) granulomatosis. A clinicopathologic study encompassing 50 years. Am J Surg Pathol 1996;20(5):519–52.
52 Monroc M, Ducou le Pointe H, Haddad S, Josset P, Montagne JP. Soft tissue signal abnormality associated with eosinophilic granuloma. Correlation of MR imaging with pathologic findings. Pediatr Radiol 1994;24(5):328–32.
53 Ando A, Hatori M, Hosaka M, Hagiwara Y, Kita A, Itoi E. Eosinophilic granuloma arising from the pelvis in children: a report of three cases. Ups J Med Sci 2008;113(2):209–16.
54 Denaro L, Longo UG, Papalia R, Di Martino A, Maffulli N, Denaro V. Eosinophilic granuloma of the pediatric cervical spine. Spine 2008;33(24):E936–41.
55 Watanabe T, Iinuma Y, Naito S, Nitta K. Eosinophilic tumor in a patient with bronchial asthma receiving pranlukast. Eur J Pediatr 2007;166(2):183–4.
56 Ladisch S, Jaffe ES. The histiocytoses. In: Pizzo PA, Poplack DG (eds) Principles and Practice of Pediatric Oncology, 3rd edn. Philadelphia: Lippincott, 1997, pp.613–31.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


