39 Treacher–Collins syndrome
Synopsis





Historical Perspective
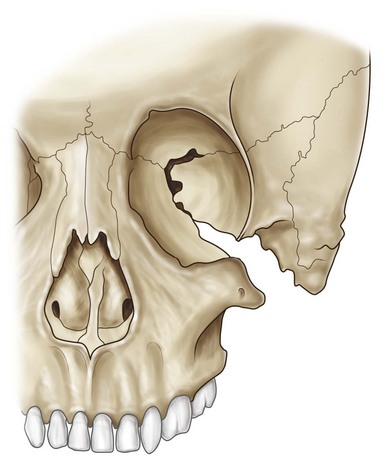
From the description by Thompson (1846)1 to that by Berry,2 Treacher–Collins (1900),3 and Pires de Lima (1944),1 this disease entity has been widely studied. The French and European literature refers to this constellation of findings as Franceschetti–Klein syndrome.4,5 Franceschetti and Klein described various details of this malformation and first called it “mandibular facial dysostosis”; they proposed a classification with three different degrees of severity: complete, incomplete, and abortive. Tessier6 described the syndrome as the bilateral confluence of 6, 7, and 8 clefts, which, depending on the degree of severity, could affect the zygoma, resulting in its hypoplasia or absence. Intelligence is usually normal (Fig. 39.1).
Basic science/disease process
Treacher–Collins syndrome, or mandibulofacial dysostosis, is a complex congenital craniofacial malformation that most strikingly involves the middle and lower thirds of the face, affecting both bony structures and soft tissues. It is transmitted by an autosomal-dominant gene of variable penetrance and phenotypic expressivity. The severity of the disease increases in successive generations.7,8 Fifty percent of the cases reported in the literature do not have a documented family history, and thus an influence of exogenous factors on expressivity of the mutation may be inferred. Advanced paternal age is considered a risk factor. These genetic anomalies cause bilateral defects in structures derived from the first and second branchial arches.
Diagnosis/patient presentation
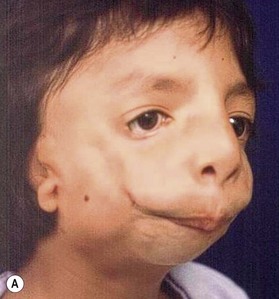
Patients with mandibulofacial dysostosis may present with all or most of the following features (Box 39.1): an inferior obliquity and shortening of the palpebral fissures, coloboma of the lower eyelids, dystopia of the lateral canthi, absence of eyelashes, and notching of the eyebrows and upper eyelids. The craniofacial skeleton is also involved: the malar bone is hypoplastic or absent, along with the zygomatic arch. The maxilla is narrow and underprojected with a high and narrow palate. The mandible is hypoplastic with a severe shortening of the ascending ramus; the condyle is severely affected as well. The chin is long and retruded; the mandibular body is short and typically features an exaggerated antegonial notch. Micrognathia of different degrees is also observed, with an anterior open bite. The nose is protruded and broad with a flattened frontonasal angle. Other clinical features include ear deformities or microtia, absence of the external auditory canal, anomalies of the middle ear, and macrostomia9 (Fig. 39.1).
Box 39.1
Characteristic clinical features of Treacher–Collins syndrome
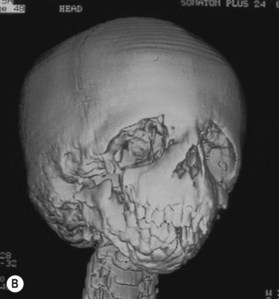
Radiographically, the Waters and posterior–anterior views, as well as frontal tomograms, show hypoplasia of the malar bones and partial or complete absence of the zygomatic arches. The shape of the orbits is abnormal secondary to the partial or total absence of the lateral wall and floor of the orbits.10
The lateral cephalogram demonstrates normal upper anterior face height with a reduced posterior face height, resulting in a vertical occlusal plane and shortening of the choanae. The sphenoethmoid angle is more acute and the angle between the anterior cranial base and the palatal plane is more obtuse. Mandibular retrognathism is present, with shortening of the ramus and the body of the mandible. The chin is long and retruded.11
The absence of the zygomatic bone is responsible for the absence of the lateral orbital rim and for the poor definition of the inferior orbital rim. For the same reason, there is no clear separation between the orbital cavity, the temporal fossa, and the infratemporal fossa. The zygomatic arches are hypoplastic or absent, and the aponeurosis of the hypoplastic temporal muscle is in direct continuity with the aponeurosis of the masseter muscle.12
According to Tessier’s classification, the zygomatic bone is absent because of the confluence of clefts 6, 7, and 8. The number 6 cleft is situated between the maxilla and the zygomatic bone, opening the infraorbital fissure. The number 7 cleft is a temporozygomatic cleft accounting for the malformations of the ears and the macrostomia. The number 8 cleft involves the frontozygomatic suture6 (Fig. 39.2). There exists a wide spectrum of phenotypic expressivity, and some clinical features are present in a less evident fashion. Likewise, there are asymmetric cases resulting from different penetrance of the deformity on each side of the face.