35 Syndromic craniosynostosis
Synopsis




Historical perspective
Over the ages, infants born with craniofacial malformations have evoked feelings of both fear and repulsion. Plato, in his Republic, suggested the elimination of malformed children, not only because they were considered a harbinger of misfortune, but also to keep the race as strong and perfect as possible.1 One of the first phenotypic descriptions of an abnormal skull shape has been attributed to Vesalius, whose observations on oxycephaly were published in 1543.2 Early treatments for abnormal skull shapes relied on the use of external molding bandages, and it was not until the 19th century that surgeons first reported attempted surgical interventions. Motivated to “unlock the brain” and relieve “mental imbecility,” Lane was among the first to treat craniosynostosis in an infant with strip craniectomies (this “relief” was short-lived, as the child died early postoperatively).3 With the recognition that sutural fusion resulted in an impairment of normal skull growth, it seemed logical that if the affected suture was removed, normal growth could resume unabated. In spite of reports of extremely high mortality rates, many surgeons continued to perform linear craniectomies for craniosynostosis throughout the last century.2 However, as experience with these strip craniectomy procedures grew, it became evident that isolated removal of an affected suture in infancy provided only limited improvement, partly because of the rapid regrowth of bridging bone. These observations resulted in the development of adjunctive techniques meant to impair reossification, such as: the creation of wider osteotomy gaps; chemical cauterization of fresh osteotomy sites; and wrapping the edges of bone with various materials.4,5 It was subsequently observed that, even in spite of surrounding the margins of freshly cut bone with polyethylene film, these bony gaps regrew fairly quickly.6 Craniofacial surgeons came to appreciate that by simply removing a section of skull (or even the entire skull!), it was not possible to create a functioning suture, and that more was needed to correct the effects of craniosynostosis.7,8 These realizations led surgeons to recognize the necessity of surgically repositioning the affected areas of the skull, in order to accomplish the restoration of normal calvarial architecture.
Through the misfortunes of war, those caring for the wounded acquired considerable experience in treating skull and facial deformities. Building on this knowledge, surgeons began to address the treatment of the facial anomalies associated with the syndromic craniosynostoses. The early treatment of the recessed midface was initially reserved until the completion of facial growth, and utilized facial osteotomies that were based on the fracture pattern lines reported by René Le Fort in the early 1900s.9,10 In 1950, Gillies and Harrison reported a using a Le Fort III-type osteotomy to advance the midface in a young adult with Crouzon syndrome; however, they were challenged by an inability to provide fixation sufficient to stabilize their advancement.11 Subsequent to this published attempt, Tessier reported his foray into treating rare facial malformations and “the monstrous deformities such as Crouzon’s and Apert’s disease.”12,13 He improved on Gillies’ Le Fort III procedure by placing the orbital osteotomies posterior to the lacrimal fossa (instead of anterior), and using interpositional bone grafts (particularly in the interpterygomaxillary region) that were secured with wire fixation. Tessier also described the use of a bandeau for skull reconstruction, which was an area that heretofore had not been addressed by plastic surgeons, as well as subperiosteal orbital osteotomies for moving the orbits. Catalyzed by these reports of paradigm shift procedures, plastic surgeons developed a renewed interest in treating the craniofacial dysostoses and other rare birth defects, and craniofacial surgery emerged as a true subspecialty.
Basic science/disease process
As the field of craniofacial genetics has matured, a more complete picture of the molecular basis for the syndromic synostoses is emerging. Today, over 150 different craniosynostosis syndromes have been described.14 The majority of syndromic craniosynostoses are the result of an FGFR-related mutations, which are primarily autosomal-dominant.15 In addition to the more commonly encountered Apert, Crouzon, and Pfeiffer syndromes, other FGFR-related craniosynostosis syndromes include: Muenke, Crouzon with acanthosis nigricans, Jackson–Weiss, and Beare–Stevenson (Table 35.1). Among the non-FGFR mutations are: Boston-type craniosynostosis (MSX2), the Philadelphia type, and Saethre–Chotzen (TWIST 1). All the FGFR-associated craniosynostoses are gain-of-function mutations, and while MSXS is also a gain-of-function mutation, the TWIST mutation represents a loss of function.16 Notably, the TWIST mutation has been reported to occur in association with an increased incidence of breast and renal cancers; however, a subsequently published multicenter Australian study has failed to support these earlier findings.17–19
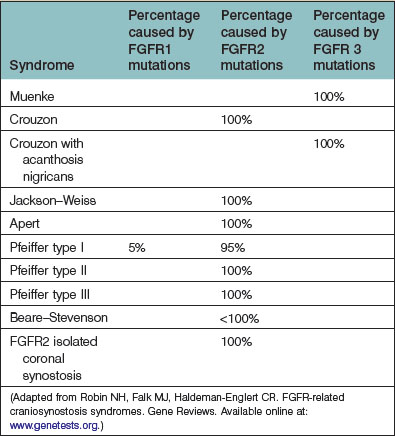
The majority of infants born with one of the syndromic craniosynostoses will present with bilateral coronal craniosynostosis, either in isolation or associated with other sutural synostoses. In addition, there may be variable effects on the development of the midface, as well as the hands and feet. It has been noted that some genes expressed in craniofacial development are also expressed in limb development.20,21 Among the most commonly encountered syndromic craniosynostoses is Crouzon syndrome, which is notable for the presence of phenotypically normal hands and feet. Pfeiffer syndrome is recognizable by the presence of enlarged thumbs and halluces, and Apert syndrome may be easily identified by the associated complex syndactylies of the hands and feet. With a fairly recent explosion of research in molecular genetics, many surgeons were hopeful that gene testing would finally provide specific and accurate diagnoses for each of the phenotypically unique craniofacial syndromes. Thus far, this has proven not to be the case. Not only can the same mutation cause different syndromes, but different mutations may be associated with the same syndrome.22,23 For example, identical mutations have been noted in individuals with Crouzon syndrome, Pfeiffer syndrome, and Jackson–Weiss syndrome, suggesting that unlinked modifier genes, or epigenic factors, play an important role in determining the final phenotype.24,25 Apert syndrome has been shown to result from at least two separate amino acid missense substitutions: Ser252Trp or Pro253Arg. The former has been reported to occur slightly more commonly (over 60% of cases) and is associated with a less severe form of syndactyly, but with an increased frequency for cleft palates.26 Moreover, within syndromes there can be significant phenotypic variability. For example, Pfeiffer syndrome has been phenotypically subclassified into three separate types: type I is described as the “classic Pfeiffer” and represents a milder presentation; type II is notable for a cloverleaf (or Kleeblattschädel) skull deformity; and type III is reserved for the most severely affected children. In spite of these differences, no specific genetic mutations have been noted to correlate with each of these described phenotypes.27
Diagnosis/patient presentation
Most physicians begin their evaluation of an infant with an abnormal skull shape by ordering a radiological study, such as a computed tomography scan, in order to ascertain the specific diagnosis. However, these studies are most often unnecessary, as the diagnosis of syndromic craniosynostoses can usually be made solely on the basis of a careful physical examination.28 Having an appreciation for how the skull is affected by sutural fusion (with growth inhibition occurring perpendicular to fused sutures and compensatory growth occurring in the remaining open sutures) enables the astute examiner to diagnose correctly which sutures are fused. Further differentiation between syndromes can be made by examination of the fingers and toes. Table 35.2 lists some of the more commonly presenting syndromes, along with some identifying phenotypic characteristics.
In early infancy, central sleep apnea (typically associated with an acquired Chiari I malformation, which results in brainstem compression) is less commonly identified as the cause for impaired ventilation, and obstructive sleep apnea is far more likely. The etiology for any observed obstructive sleep apnea may be multifactorial, but is most often directly related to mid facial hypoplasia. This hypoplasia elevates the palate, which correspondingly reduces the size of the nasal airway. This resultant diminished nasal airway is different from a true choanal atresia, which is caused by a congenital persistence of tissues separating the nose form the mouth. The reduced nasal aperture associated with syndromic craniosynostosis is best left untreated because of the dismal success rates that have been observed following any early surgical intervention.29 Other causes for airway obstruction include: tracheomalacia, tracheal stenosis (especially with type II Pfeiffer syndrome), and gastroesophageal reflux (antireflux medication should be considered for all syndromic infants).30 Feeding issues are commonly seen in conjunction with airway obstruction, and associated neuromuscular immaturity may result in silent aspiration; therefore, feeding evaluations are often useful. When obstructive sleep apnea is identified, more conservative treatments include use of continuous positive airway pressure masks and tonsillectomies. Some surgeons have reported performing frontofacial advancements in infancy; however, there is currently no evidence to support these early interventions, and such treatments may be considered as falling outside the current mainstream of care.31,32 On the other hand, temporary tracheostomies have been attributed to lowering mortality rates for the more severe patterns of syndromic craniosynostosis, and should be considered in all infants and younger children who have failed the more conservative therapies.30 Typically, the incidence for airway compromise can be expected to increase with age, as the mid facial hypoplasia becomes more progressive.
In addition to airway management, the second critical area of focus for any child with syndromic craniosynostosis is the avoidance of raised intracranial pressure. As with airway issues, the potential for raised intracranial pressure increases over time. Unfortunately, the clinical determination of elevated intracranial pressure is an extremely challenging endeavor. There have been no published studies evaluating either the specificity or sensitivity of any of the often-cited clinical signs for raised intracranial pressure. Currently, it is unknown exactly how high, or for how long intracranial pressure needs to be elevated before there are irreversible adverse effects on cognitive function. Nevertheless, some attempts to determine the presence of potentially elevated intracranial pressure must be made. There are studies using direct measurement techniques, which suggest that when more than one suture is fused (even when there is a normal skull volume), intracranial pressure is more likely to be elevated.33–35 Given the invasive nature of obtaining direct intracranial pressure measurements, it is not reasonable to perform serial testing for elevations in intracranial pressure in every child with syndromic craniosynostosis. Instead, secondary assessments for intracranial pressure are used, such as: the physical assessment of fontanels or other skull defects (to assess dural tension), serial head circumference measurements (to look for changes along the growth curves), fundoscopic examinations (to assess for papilledema), reversed visual evoked potentials, and magnetic resonance imaging scanning (to monitor for decreasing ventricular size, enlargement of the optic nerves, or progressive tonsillar herniation).35–40 Some believe that chronic elevations in intracranial pressure may result in observable changes in the skull radiographs, such as imprinting (“copper-beaten,” and/or thinning of the bone); however, there are no supportive studies to confirm this relationship, nor does this finding appear to correlate with intelligence.41 Although it may seem intuitive that progressive skull malformations might correlate with increased intracranial pressure and developmental issues, this relationship has thus far not been supported by retrospective single sutural synostosis studies.42,43
In addition to the need for evaluating a child for potential airway compromise and raised intracranial pressure, additional anomalies must be considered. The association between cerebellar tonsillar herniation and syndromic craniosynostosis was first described over 30 years ago; since that time, studies have shown that these Chiari malformations are an acquired defect, and are likely exacerbated by ventriculoperitoneal shunting.44–46 Chiari malformations have been observed to occur infrequently in Apert syndrome, often in Crouzon syndrome, and almost always in the more severe presentations for Pfeiffer syndrome.30,37,47 It is important to screen all children for this condition with routine magnetic resonance imaging, because, when symptomatic, Chiari malformations may lead to central sleep apnea, disordered swallowing, syringomyelia, or even potentially fatal central sleep apnea. In addition to Chiari malformations, children need to be followed for the potential development for hydrocephalus, which is often not diagnosed until after a skull expansion procedure has been performed.
Another important concern in children born with syndromic craniosynostosis is exposure-related visual loss. As a result of altered orbital growth, severe proptosis may occur, resulting in impaired lid coverage and corneal scarring. Some infants with syndromic craniosynostosis (primarily with Apert syndrome) may also have submucous, or complete clefts of the secondary palate. There is also a higher incidence of cardiac anomalies among the syndromic craniosynostoses, particularly atrial and ventricular septal defects.48 Malrotation of the gut appears to be more common in Pfeiffer syndrome, and should be radiologically evaluated with an upper gastrointestinal series with a small-bowel followthrough, when indicated.30
Patient selection
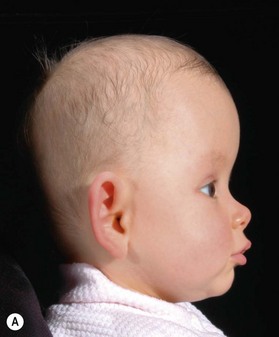
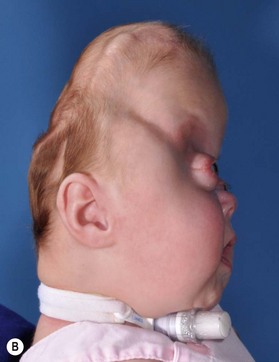
Identification of the ideal time to operate on a child, and determining which operation should be performed, cannot be decided on a syndrome-by-syndrome basis. Instead, it is better to consider the child’s phenotypic presentation. In this regard, it is useful to recognize that the syndromic synostoses actually represent a continuum of birth defects. At one end of this syndromic craniosynostosis spectrum would be the isolated bilateral coronal craniosynostoses (without any associated mid facial hypoplasia, and only minimally impaired cranial growth), and at the other end would be the severely constricted Kleeblattschädel skull deformities and complete pansynostoses (with extremely deficient mid facial growth, and associated severe airway anomalies) (Fig. 35.1). Appreciating where each child falls on this continuum allows surgeons to apply treatment algorithms specific to the problems encountered.
The timing for the initial surgical intervention for any of the craniosynostoses has not been specifically studied. The two goals for treating the affected skull of any child with syndromic craniosynostosis are to normalize appearance, and to prevent sustained elevations in intracranial pressure (sufficient to impact cognition). Implicit among these goals is to accomplish them as safely, and with as few procedures, as possible. Studies examining long-term calvarial growth following single sutural corrections have shown that growth is not normal postoperatively.49,50 Considering the rapid growth of the brain in infancy, delaying surgical intervention has the benefit of achieving a better longer-term result. This is partly because early surgical intervention must contend with the reduced mechanical rigidity of the thin infant skull that impairs the ability to achieve any significant enlargement of the calvaria. Moreover, earlier surgical intervention has the disadvantage of the rapidly growing brain quickly occupying any achievable increase in intracranial volume. Conversely, delaying surgery might potentially result in a preventable visual or developmental loss.36,38 Studies measuring intracranial pressure in children with craniosynostosis suggest that elevated pressure is more likely when multiple sutures are fused, that elevated pressures may inversely correlate with mental function, and that surgery can successfully reduce intracranial pressure.33–35 However, more important than treating raised intracranial pressure is the prevention of avoidable developmental delays; this particular parameter is something that is much harder to measure. Some studies have suggested that earlier surgery (under 1 year of age) is associated with higher IQ scores.51,52 Nevertheless, these retrospective studies may have been influenced by selection biases (i.e., well-educated parents may be more likely to bring their child to a physician earlier for treatment).
Treatment/surgical technique
Of all the congenital craniofacial anomalies, treatment of the syndromic craniosynostoses can present some of the greatest challenges. Although the vast majority of published literature has been retrospective and anecdotal, much has nevertheless been learned from observation. The aforementioned goals of treatment, which are to normalize both function and appearance, need to be accomplished as safely and with as few operations as possible. The management of the syndromic craniosynostoses requires a team of dedicated specialists, and is ideally reserved for those centers that have both a focused interest in these complex problems and sufficient expertise to provide safe and efficient care. Underscoring the challenges of treating the more severe syndromic craniosynostoses are the significant mortality rates that have been described within this particular subpopulation (reportedly as high as 66–85%).53,54 The timing for skull surgery, and the selection of the best operative procedure, varies with the child’s phenotype. At the milder end of the syndromic craniosynostosis spectrum, presenting with a more moderate degree of brachycephaly (especially when there is a split metopic or sagittal suture that offers some degree of decompression), the author will delay surgery until the open bony defects have closed, or the dura begins to become tense. As the degree of skull deformity progresses along the severity continuum, progressive turricephaly will be seen. If allowed to progress beyond a moderate stage, an increased skull height can be exceedingly difficult to correct later, therefore making significant turricephaly a relative indication for earlier surgical intervention.
The surgical procedure for any child falling along the mild to moderate end of the syndromic craniosynostosis spectrum consists of enlargement of the anterior fossa. The goal of surgery is to increase intracranial volume by bringing the supraorbital bandeau as far forward as possible, while simultaneously lowering any anterior turricephaly. Surgery performed in children under 9 months of age is more challenging because of the inherent weakness of the thin, relatively immature skull; as a result, any small degree of advancement obtained at this early age will be quickly eradicated by the rapidly growing brain (clinically evident by the rapid reappearance of brachycephaly). Perhaps it is for this reason that some surgeons have advocated beginning with a posterior decompression.55,56
However, there are also a number of compelling reasons that argue against early posterior skull surgery in children with syndromic synostosis. To begin with, acquired Chiari malformations, although relatively uncommon for Apert syndrome, have been reported to occur in 70% of children with Crouzon syndrome and up to 100% of children with the more severe Pfeiffer presentations.30,37,47






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


